1- Épidémiologie :
– Le risque de développer une SEP dépend de la prévalence de région où on a passé les 15 premières années de sa vie.
– Il existe un gradient nord-sud prévalence plus élevée au nord.
– Prédominance féminine 60 % ; l’âge moyen de début est de 30 ans. 70 % entre 20 et 40 ans
2- Physiopathologie :
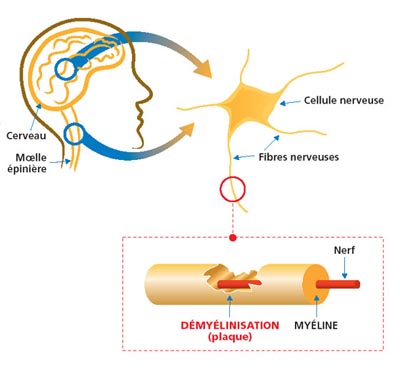
– Plaques de démyélinisation disposées sans ordre dans la substance blanche et de façon élective prés du LCR : périventriculaire et centre ovale des hé-misphères cérébraux, nerf optique, le tronc cérébral, le cervelet et la moelle épinière.
– Au début inflammation (infiltrat mononucléé lymphoplasmocytaire, périvasculaire) puis désintégration de la myéline (phagocytée par le macrophages).
– La démyélinisation respecte relativement les axones : c’est la dissociation myélino-axonale.
– La remyélinisation est plus ou moins complète, accompagnée d’une gliose astrocytaire réactionnelle.
– Souffrance axonale (dés le début de la maladie) : aboutit à un handicap permanent. Coexistence de plaques d’âges différents (dissémination dans le temps) d’une façon diffuse dans le SNC (dissémination dans l’espace).

– La substance grise et le système nerveux périphérique sont généralement respectés
– Les nerfs crâniens peuvent être atteints dans leur trajet intranévraxique (exp : V et VII)
– Une atteinte axonale secondaire est possible après plusieurs poussées expliquant les séquelles entre les poussées.
– Les phénomènes paroxystiques sont liés à des phénomènes de membrane (bloc de conduction) et non à la démyélinisation. Ces blocs de conduction sont aggravés par la chaleur et l’acidose.
– Sur le plan étiologique, beaucoup de facteurs sont incriminés : virus (Ac antirougeole++) ; environnement (durant l’enfance) ; auto-immunité : synthèse intrathécal d’Ig polyclonales ; réduction des lymphocytes T CD8+ au moment des poussées (qui sont élevés lors de la rémission). Rôle de TNFα et de l’IFNγ. Production d’Ac antimyéline. Il existe une prédisposition génétique mais les formes familiales sont rares.
3- Clinique :
– Evolution marquée par des poussées régressives (forme rémittente, la plus fréquente).
– Troubles sensitives fréquents et parfois caractéristiques. Paresthésies ; dysesthésie de contact ; signe de Lhermitte qui est un équivalent d’une atteinte cordonale postérieure avec sensation de décharge électrique brève déclenchée par la flexion de la nuque, descendant le long du dos et des membres in-férieurs.
– Atteinte motrice (pyramidale et cérébelleuse) :
* L’atteinte de la voie pyramidale est fréquente (80% après 5 ans d’évolution) ; différents formes cliniques (monoplégie, hémiplégie…) syndrome py-ramidal (voir le cours)
* Syndrome cérébelleux (50 % des cas) statique et cinétique avec souvent un tremblement intensionnel souvent invalidant
– Atteinte du nerf optique : presque constante, souvent infraclinique avec PEV seuls altérés. Elle réalise une neuropathie optique rétrobulbaire. 22% des ESP débutent par cette atteinte. Elle se traduit par une baisse brutale de l’acuité visuelle, unilatérale, avec douleurs orbitaires. Parfois incomplète avec altération de la vision de couleurs (dyschromatopsie rouge-vert) et scotome central. Le fond d’œil est normal au début, secondairement apparaît une pâleur du segment temporal de la papille.
– Atteinte du tronc cérébral :
* Troubles de l’oculomotricité : la diplopie est fréquente, le plus souvent liée à une ophtalmoplégie internucléaire ou OIN (III <=>VI).
En cas d’OIN, l’œil en abduction (VI) n’est plus suivi par l’œil controlatéral (III), d’où une diplopie horizontale, alors que la convergence des yeux est possible. Une OIN bilatérale est très évocatrice du SEP.
* Névralgie symptomatique du V (trijumeau) : douleurs fulgurante dans les territoires du V, il existe une hypoesthésie et une abolition des réflexe cornéen du même côté (éliminant un névralgie essentielle du V).
* Paralysie faciale de type périphérique
* Syndrome vestibulaire de type central (dysharmonieux, vertige fugace, nystagmus multidirectionnelle).
* NB : la surdité est rare mais une altération infraclinique du PEA est fréquente.
– Troubles sphinctérien et sexuels : fréquentes, quasi constante après une évolution de plus de 10 ans. Miction impérieuse et pollakiurie, dysurie, résidu post-mictionnel. Impuissance et frigidité.
– Troubles psychiques : thymique (dépression), troubles intellectuels portant sur l’attention, la mémorisation, plus rarement démence sous corticale.
– Autres signes :
* Signes généraux : asthénie très fréquente,
* Manifestations paroxystiques : crises toniques brèves ; dysarthrie paroxystiques ; névralgies (sensibles au Tégrétol : carbamazépine).
* Signes importants : aggravation clinique ou apparition de nouvelles manifestations liée à un effort physique soutenu, à une forte chaleur, à un bain chaud ou au cours d’un syndrome fébrile (bloc de conduction).
* Signes négatifs : pas d’atteinte corticale (pas d’aphasie ni apraxie, épilepsie exceptionnelle) ; par d’atteinte du SNP ; pas de signes extrapyramidaux ; hémianopsie latérale homonyme exceptionnelle.
* Anomalies pupillaires possibles : signe d’Argyll-Robertson (anisocorie avec abolition du réflexe photomoteur et conservation de l’accomodation-convergence) ; phénomène pupillaire de Marcus-Gunn (mydriase paradoxale à l’éclairement de l’œil atteint par une neuropathie optique rétrobulbaire
4- Examens complémentaires :
– Au examen n’es spécifique de la sclérose en plaque.
– FNS normale, pas de syndrome inflammatoire, VS normale.
– LCR : Hypergammaglobulinorachie évocatrice (> 12 % de la protéinorachie) due à une synthèse intrathécale d’IgG sans spécificité antigénique connue. Elle est de distribution oligoclonale (bande oligoclonale), absente du sérum. Une pléïocytose lymphocytaire (< 50 éléments/mm3) peut être observée ; LCR normal n’élimine pas le Dc ; absence de corrélation entre anomalies du LCR et la gravité des poussées.
– IRM : dissociation anatomoclinique ; en sus- et soustentoriel et dans la moelle ; dans la substance blanche et surtout périventriculaire. En T1 : hyposignal prenant le gadolinium en cas de jeune plaque (< 3 mois). En T2 : zones d’hypersignal. Pas d’effets de masse. Elles ne sont pas spécifiques du SEP. Elles sont variables d’un examen à l’autre. Une atrophie des corps calleux est fréquemment observée. La prise de gadolinium est un marqueur de d’évolutivité de la maladie. Le scanner est moins sensible que l’IRM (les plaques récentes sont isodenses et prennent le contraste ; les plaques anciennes sont hypodenses et ne sont plus rehaussées par le produit). A un stade plus avancé -> dilatation ventriculaire.
– Potentiels évoqués multimodaux (PEA, PEV, PES, PEM) : montrent les lésions mêmes infracliniques ; multifocalité des lésions
5- Diagnostic positif :
– Dissémination dans le temps ; dissémination dans l’espace, synthèse intrathécale d’Ig (LCR) ; absence d’autres causes (la SEP reste un Dc d’élimination)
– Deux poussées cliniques avec deux lésions anatomiques distinctes suffisent à confirmer le diagnostic de SEP.
6- Évolution :
– Le début peut être monosymptomatique dans 50% des cas
– L’évolution par poussées régressives est fréquente (80%) ; c’est la forme rémittente. Qui est caractéristique de la SEP
– La poussée est définie par l’apparition de nouveau signe ou l’aggravation d’un signe préexistant, en dehors de tout facteur intercurrent, durant plus de 24 heures et survenant à plus d’un mois de la dernière poussée.
– Une rémission est définie par une amélioration persistante des signes durant au moins un mois.
– La régression des signes est de moins au moins complète, laissant des séquelles entre deux poussées
– Formes secondairement progressives : après 10 ans d’évolution, la moitié des patients présenteront une forme progressive définie comme une aggravation progressive des troubles neurologiques pendant au moins 6 mois.
– La forme primitivement progressive (15%) est essentiellement médullaire ; après 40 ans ; sans prédominance féminine
– Troubles de la marche : après 6 ans d’évolution
– Patient confiné à son domicile : après 18 ans d’évolution
– Médiane de survie : 35 ans. Décès par complication de décubitus ; infection urinaire
– Facteurs de mauvais pronostic : début tardif, formes progressives, intervalle court entre les deux premières poussées, détérioration intellectuelle.
7- Traitement :
– Corticoïdes : traitement symptomatique au moment de la poussée ; permettent de réduire la durée de la poussée ; ne préviennent pas les poussées ultérieurs ; n’influence pas le cours de la maladie.
– Antispastiques : baclofène, dantrolène
– Immunosuppresseurs : (traitement de fond) sont réservés aux formes sévères. Azathioprine (forme sévères à poussées) ; cyclophosphamide (formes secondairement progressives).
– Immunomodulateurs : (traitement de fond) ; interféron ß (IFN ß) qui agit en s’opposant aux effets toxiques des l’IFNγ et du TNFα et en activant les lymphocytes suppresseurs.
Vous devez être connecté pour poster un commentaire.