 Introduction:
Introduction:
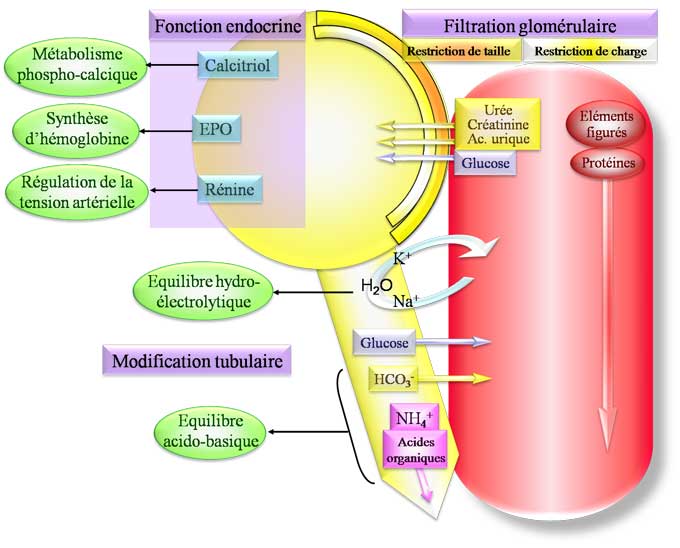
The vital role of the kidneys is intimately linked to their function in the homeostasis of the internal environment, thus protecting the cells from the consequences of environmental variations of the organism. The kidneys eliminate the terminal metabolic products (urea, protons, uric acid, creatinine, bilirubin, etc.), a large number of exogenous substances such as certain drugs and food additives, and an adjusted amount of water and electrolytes.Tubular functions play a central role in this day-to-day adjustment of the final urine composition, of which they constitute, with a few exceptions, the limiting step.
It is only at a late stage of renal failure that the decrease in glomerular filtration rate (DFG) becomes sufficient to alter the hydroelectrolytic homeostasis capacities by itself. However, tubular functions are frequently altered at an early stage of glomerular nephropathy and these alterations are of predictive value in the course of chronic renal failure.
Indeed, it has been shown that the rate of decrease of GFR during glomerular nephropathies was better correlated with the importance of tubulo-interstitial functional and morphological lesions (tubulo-interstitial fibrosis or tubular atrophy) than to the glomerular lesions themselves .
The study of tubular functions thus finds two main indications:
• research and characterization of a primary deficit of one or more tubular functions;
• detection of early alterations in tubular functions during glomerular renal disease.
A congenital or acquired primary tubular deficiency should be distinguished from non-specific tubular disorders of tubulo-interstitial nephropathy. In the presence of renal insufficiency, the exploration of tubular functions must therefore determine whether the abnormalities observed are related to the degree of renal insufficiency or whether they are disproportionate to the degree of renal insufficiency, suggesting primary tubular damage.
Physiological reminder:
Functional segmentation of the renal tubule:
Before considering the study of tubular functions, some reminders of renal physiology should be made, which the reader may complete by reading specific works in French or English.
The tubule is subdivided into three functional parts: the proximal tubule, the Henle’s loop, and the distal nephron, which extends from the distal tubule distal to the collecting duct.
The proximal tubule reabsorbs almost all of the amino acids and glucose, 70% of water, sodium, phosphate and 80% of the bicarbonate filtered by the glomeruli. It is also the main site of reabsorption of calcium, reabsorption / secretion of anions and organic cations, and participates in the reabsorption of magnesium. Finally, the proximal tube is the site of an activity of endocytosis and of intense lysosomal degradation. Generally, a partial or total proximal alteration (Fanconi syndrome) of the tubular functions significantly alters the maximum reabsorption capacity (Tm) of one or more electrolytes and is accompanied by tubular proteinuria.
The thin descending branch and the thin, upward branch of the loop of Henle reabsorbate 20 to 30% of the sodium, 15 to 20% of the bicarbonate, 30% of the calcium and 70% of the magnesium filtered by the glomerulus. Henle’s loop is the site initiating the formation of the corticopapillary gradient, which relies on interstitial accumulation in the medullary external and internal osmoles (urea, sodium and potassium) and NH 3 / NH 4+ . Sodium chloride (NaCl) reabsorption in the macula densa, located at the end of Henle’s Loop, is the initial stage of tubuloglomerular feedback that allows DFG to be adjusted to maintain constant fluid flow and of NaCl delivered to the distal nephron. Henle’s loop thus determines the environment of the cells of the more distal segments by adjusting not only the composition and the flow rate of the fluid delivered to these segments but also the composition of the interstitial fluid which surrounds them. An alteration of the tubular transport at this site has an important effect on the hydroelectrolytic state by qualitatively and quantitatively altering the tubular reabsorption capacities, as illustrated by the major hydroelectrolytic loss of the Bartter syndrome.
The distal nephron re-absorbs less than 10% of the filtered charge in water and electrolytes, but is the site of the adjustment of the balances. An alteration at this site, illustrated by Gitelman syndrome or pseudohypoaldosteronism type 1, has less quantitative than qualitative consequences and can lead to severe electrolyte disorders without significantly altering the maximum tubular reabsorption capacities of sodium and bicarbonate, or the power of concentration of urine.
Renal tubular functions:
Proximal and distal endocytosis:
Endo / exocytosis is a complex process that allows cells to capture exogenous macromolecules to degrade them in lysosomes or to deliver neosynthesized proteins to the plasma membrane or in extracellular media. In the renal tubule, endocytosis appears as one of the multiple facets of, or rather, tubular epithelial functions. In the proximal tubule, the endocytosis of the filtered proteins indirectly allows to preserve the essential amino acids of the organism and to purify the tubular fluid of biologically active peptides resulting from the glomerular filtration. Throughout the nephron, endo / exocytosis mechanisms are involved in the regulation of the expression of transport proteins at the plasma membrane.In the intermediate cells of the collecting channel, they could play a protective role with respect to microcalcifications and bacterial invasions. There is no specific exploration of this function, the proximal deficit of which is indirectly assessed by quantitative and especially qualitative analysis of urinary excretion of proteins, with preferential excretion of low molecular weight proteins ( a 1 and b 2 microglobulins) relative to albumin.
Tubular reabsorption and secretion:
The urinary excretion of each substance is the result of three processes: its glomerular filtration; its tubular reabsorption and secretion. The formation of the urine begins with the filtration of a volume close to 170 l / 24 hours, whose composition is very close to that of the plasma liquid. Most substances that are to be eliminated from the body are not reabsorbed (such as creatinine) or reabsorbed and secreted (like some drugs) and, therefore, are eliminated in large quantities in the urine.
Conversely, electrolytes such as sodium, chlorine and bicarbonate ions are very strongly reabsorbed and their fractional excretion is very low. If reabsorption is generally quantitatively greater than tubular secretion, tubular secretion can play a decisive role in the adjustment of the final quantity excreted in the urine. Thus, renal adaptation to high potassium intakes consists of stimulation of distal potassium secretion and not a partial inhibition of its proximal reabsorption.
Regulation of blood volume:
The kidneys of a healthy subject filter daily more than 25,000 mmol of NaCl per 24 hours. Urinary excretion of sodium in the final urine is only about 150 mmol / day, or less than 1% of the filtered feed, and reflects the sodium dietary intake.
The regulated value of the sodium balance is the effective blood volume, defined by the arterial blood volume. The decrease in effective blood volume (whether linked to true hypovolemia, decreased cardiac output or decreased peripheral resistance) leads to stimulation of antinatriuretic factors (sympathetic system, renin-angiotensinaldosterone axis) which stimulate reabsorption tubular renal NaCl and decreased sodium excretion.
Control of the acid / base state:
In the normal subject, the extracellular pH is maintained substantially constant (between 7.37 and 7.43), as is the cellular pH, the value of which, generally lower, varies according to the tissue considered from 6.8 to 7.2. The kidney removes, in the form of NH 4+ and titratable acidity, the charge to fixed food acids. It filters daily more than 4,500 mmol of bicarbonate which it must reabsorb completely, under penalty of inducing metabolic acidosis by renal loss of bicarbonate.
Finally, in the event of an exarenal loss of bicarbonate or exogenous acid supply, the kidney is capable of generating NH 4+ and bicarbonate, allowing to restore the buffer stock of the organism that transiently allowed to buffer the charge acid and excreting the acidic charge essentially as ammonia.
Regulation of the water balance:
Physiological drinking and diet expose the body to major alterations in the body’s fluid and osmotic content. The role of the kidney is to eliminate the amount of water and osmoles appropriate to maintain a water and osmotic content of the body. The kidney outputs of water are the major target of regulating the body’s water outflows, mainly under the control of the antidiuretic hormone. Stable diuresis is equivalent to the exogenous water inputs it reflects. The integrity of the regulation of the water balance rests on three elements that must be evaluated in parallel:
• antidiuretic hormone secretion and thirst, which must be stimulated by hyperosmolarity and inhibited by hypoosmolarity;
• kidney intrinsic urinary concentration / dilution abilities;
• osmotic inputs, which modulate the minimum and maximum water excretion capacities.
Renal Tubal Explorations:
Principles of exploration:
In most cases, tubular explorations are motivated by a hydroelectrolytic disorder that suspects a selective or nonselective primary renal loss (or retention) of water or electrolytes (renal losses of bicarbonate, glucose, phosphate, sodium, potassium , calcium or magnesium).
Simple tests carried out on an outpatient basis can give indications on the alterations of certain tubular functions. This analysis assumes the normal response to a decrease or, on the contrary, to a sudden increase in water, mineral and electrolyte intakes.
The study of the tubular behavior of a solute consists of measuring the filtered amount (the product of the ultrafiltrable concentration of the solute and of the GFR) and that excreted in the urine during the acute administration of this substance. This type of test makes it possible, for example, to determine the Tm of glucose or bicarbonate during the analysis of the proximal tubular functions or to characterize the tubular behavior of other solutes such as calcium or magnesium.
In order to detect and / or characterize a renal tubular adaptation abnormality, especially when there is no hydroelectrolytic disorder in the basal state, it is often necessary to test the renal response to “metabolic stress”. The three most commonly used tests are:
• the acid load test, which evaluates the function of distal acidification of the urine;
• the water restriction test with administration of 1-desamino-8-D-arginine vasopressin (dDAVP), which estimates the concentration of urine;
• the aqueous charge test, which estimates the dilution power.
These tests can be supplemented by pharmacological tests in order to locate the tubular abnormality (s) and to determine their mechanisms. This aspect is illustrated, for example, by studying the natriuretic response to various diuretics as a tool for localizing the lack of NaCl reabsorption during Bartter / Gitelman syndromes or the lack of reabsorption of the divalent cations characterizing certain hereditary tubulopathies.
Analysis of hydroelectrolyte disorders and / or mineral metabolism:
Renal losses of potassium and / or sodium chloride:
Renal sodium loss can be easily demonstrated by comparing the sodium excretion of sodium measured on a sample of urine or in the urine of 24 hours with the state of the extracellular volume. In the presence of a table of contraction of the extracellular volume of extrarenal origin, the normal response of the kidney, under the influence of secondary hyperaldosteronism essentially, is to reabsorb the quasitotality of the sodium filtered by the glomerulus. In this case, urinary sodium excretion is collapsed, less than 10 mmol / l in a sample or 20 mmol / 24 hours.
Hypokalemic sodium chloride kidney failure:
The simultaneous presence of clinical hypovolemia and / or secondary hyperaldosteronism (elevation of renin and aldosterone beyond any treatment capable of influencing their secretion) and of a natriuresis greater than 30 mmol per 24 hours allows to assert the existence of a renal loss of NaCl. In intermediate situations (sodium urinary excretion between 20 and 30 mmol / 24 hours), extracellular dehydration can first be corrected by bringing NaCl and then imposing a restriction of NaCl intake for a few days (feeds less than 20 mmol per 24 hours). The normal kidney adapts its urinary excretion of sodium to the intake (natriuresis less than 20 mmol per 24 hours) in 2 to 4 days, which is not the case for a sodium renal loss where the kidney adapts over a much longer period, explaining the onset of extracellular dehydration. This is the case for specific spinal cord injuries (polycystic kidney disease, chronic pyelonephritis, obstructive uropathy, etc.), but also chronic renal insufficiency, especially in the context of hyporeninemia-hypoaldosteronism syndrome.
A kaliuresis less than 20 mmol / 24 hours contemporary with a hypokalaemia signs the extrarenal origin of this hypokalaemia.
Conversely, a kaliuresis greater than 40 mmol per 24 hours signs the renal origin of this hypokalaemia. Note that there is an area of uncertainty that justifies repeating this exploration after oral or intravenous administration of potassium, but before total correction of this hypokalaemia.
Once the renal loss is established, it is necessary to analyze its mechanisms and in particular to investigate whether this hypokalemia associates with a renal or extrarenal loss of NaCl. If kaliuresis is evaluated on a sample, it is preferable to calculate the transtubular potassium gradient (GTTK), which takes into account the patient’s antidiuretic state and therefore the elevation of urinary concentration of potassium related to the concentration of urine.
The association of a renal loss of NaCl with hypokalemic alkalosis of renal origin suggests a decrease in sodium reabsorption in a segment located upstream of the collecting duct: broad ascending branch or distal circumvented tubule. This loss of NaCl may be related to the use of diuretics capable of specifically inhibiting sodium reabsorption in these segments: diuretics of the furosemide loop for the broad ascending limb or thiazide diuretics for the distal circumferential tube. Similar charts are observed in congenital hereditary pathologies: Bartter and Gitelman syndromes. The five subtypes of Bartter’s syndrome described to date correspond to genetic defects involving proteins directly or indirectly involved in NaCl cell transport in the broad ascending limb of Henle’s loop, while Gitelman’s syndrome is generally secondary to a gene inactivation of the thiazide-sensitive cotransport NaCl or, more exceptionally, of the basolateral chloride channel ClCKb. These syndromes can be differentiated by the precocity of the signs, the presence of an antenatal hydramnios, a lack of urine concentration, nephrocalcinosis and / or hypercalciuria (characteristics of Bartter’s syndrome) or a hypomagnesemia which is more severe in Gitelman syndrome.
Chloruria is preserved during the intake of hypokalemic diuretics and in Bartter / Gitelman syndromes. It makes it possible to differentiate these states from the loss of sodium and potassium induced by profuse vomiting associated with a chloruria of less than 10 mmol / l. In this latter situation, the extra-alkaline loss of hydrochloric acid corresponds to a loss of chlorine associated with a sudden addition of bicarbonate, which the kidney eliminates in association with sodium and potassium. Chlorinated depletion stimulates renal reabsorption of chlorine, as evidenced by collapsed chloruria (less than 10 mmol per 24 hours) while natriuresis is conserved and alkaline urinary pH greater than 7.
At the end of vomiting, the previously established hypovolemia stimulates renal sodium reabsorption and natriuresis becomes adapted to hypovolemia (natriuresis less than 10 mmol / l or 20 mmol per 24 hours). On the other hand, secondary hyperaldosteronism, which stimulates distal potassium secretion, explains that kaliuresis remains inappropriate for hypokalemia, which was previously established until the blood volume is corrected.
The repetition of measurements of natriuresis and chloruria is therefore an important element in the etiological diagnosis of renal hypokalaemia. Natriuresis is consistently unsuitable for hypovolemia in Bartter and Gitelman syndromes. On the contrary, diuretics and intermittent vomiting lead to periods of sodium renal loss, which are separated by periods during which diuretic or vomiting is stopped, allowing the renal adaptation to the hypovolemia demonstrated by periodically adapted natriuresis.
Hyperkalaemic sodium chloride kidney failure:
A loss of NaCl associated with hyperkalemia and metabolic acidosis should prompt a primary defect (mineralocorticoid insufficiency) or secondary defect of aldosterone secretion in the context of hyporenemia hypoaldosteronism. In this context, the exceptional presence of high concentrations of renin and aldosterone resembles a type 1 pseudohypoaldosteronism. In pseudohypoaldosteronism type 1, renal NaCl loss and hyperkalaemic acidosis are associated with elevated plasma renin concentrations and aldosterone, indicative of renal resistance to the action of aldosterone. These rare conditions may be secondary to inactivating mutations of the mineralocorticoid receptor gene or that of the EnaC channel responsible for sodium reabsorption in the collecting channel.
Renal response to metabolic acidosis:
The analysis of the renal response to metabolic acidosis is more complex. The first step consists in calculating the plasma anionic hole (TAP = [Na] p [K] p – [CI] p [HCO3] p, normal: 17-21 mmol / l). In the presence of hyperchloremic acidosis (normal TAP), renal response analysis allows the differentiation of acid overload with normal renal function from the primary decrease in the excretion capacity of H + ions, defining tubular acidosis. In extrarenal acidosis, the normal kidney maximally adapts its renal excretion capacity to H + and the urinary excretion of NH 4+ is therefore
still high, greater than 70 mmol / 24 hours, indicating an appropriate response of the kidney. In renal acidosis, the excretion of NH 4 + is less than or equal to 40 mmol of H + ions per 24 hours. Since urinary NH 4+ measurement is not yet common practice, it has been proposed to estimate ammonia by calculating the urine anion hole (TAU). The TAU is equal to the difference between anions and cations indosed in urine (TAU: Na + K – Cl, the quantity of urinary HCO 3 being considered quantitatively negligible when the urine pH remains below 6.5). The principle is that increased excretion of NH 4 results in increased excretion of indosed cations and chlorine. The greater the ammoniuria, the more the TAU decreases, until negativer. Urinary excretion of NH 4+ is approximately 40 mmol / 24 hours in a normal renal function patient without acidosis and western diet, and physiological TAU is positive (approximately 30 mmol / 24 hours). In the case of extrarenal metabolic acidosis, the progressive increase of urinary NH 4 to a value greater than 70 mmol / 24 hours (the other anions and indosed cations not varying significantly) explains why the anionic urinary hole diminishes and becomes negative. During renal acidosis, there is no increase in urinary excretion of NH 4 and urinary TAU remains positive. The limit of this approach is that the measurement of chloruria is sometimes more difficult to obtain routinely than that of ammoniuria.
The physiopathological analysis of tubular acidosis requires dynamic tests: in practice, a bicarbonate loading test, possibly supplemented with a furosemide test. Finally, the absence of metabolic acidosis in the basal state does not exclude a defect of distal acid excretion (partial distal acidosis) which requires, to be objectified (or excluded) the realization of an acid charge test.
Chronic renal failure is associated with the progressive onset of metabolic acidosis with increased plasma anion gap, reflecting the plasma accumulation of partially dissociated anions at plasma pH. In the presence of renal insufficiency, the appearance of metabolic acidosis without parallel elevation of the plasma anionic hole should suggest an acidosis of extrarenal origin or the presence of superadded tubular proximal acidosis.
Renal response to variation in plasma osmolarity (renal regulation of the water balance):
Any analysis of the water balance must begin with the analysis of the state of hydration intra- and extracellular.Natremia is a reflection of effective osmolality, except when there are significant amounts of osmotic substances other than sodium. Measurement of plasma osmolarity and osmotic hole calculation, the difference between the measured plasma osmolarity and the calculated osmolarity (Å ~ 2) may reveal an increased osmotic hole (greater than 10 mOsm / kg of water) . Urea and blood glucose should then be measured and the notion of chronicity of a possible hyperglycemia should be evaluated in order to clarify whether or not the osmole in question is osmotically active. Thus, an elevation of the osmotic hole by elevation of non-osmotically active urea is common in chronic renal failure.
Hypotonic hyponatraemia necessarily corresponds to intracellular hyperhydration, which must result in a dilution state.In this situation, urinary osmolarity greater than 150 mOsm / kg of water indicates an inappropriate antidiuretic state, while an osmolarity of less than 150 mOsm / kg of water indicates a water intake exceeding normal water excretion capacities or altered by low osmotic inputs. Conversely, an effective hyperosmolarity demonstrates intracellular dehydration, which must lead to water retention. A urinary osmolarity of less than 850 mOsm / kg of hypernatremia shows an alteration in the urine concentration, while a urinary osmolarity of more than 850 mOsm / kg, which is contemporaneous with hypernatremia, indicates an extreme hydric loss or hypertonic intake exceeding normal kidney abilities. However, this last judgment criterion must be adjusted to the degree of the polyuria possibly associated, which in itself alters the power of concentration.
Exploration of the tubular behavior of one or more solutes:
Certain substances such as glucose and bicarbonate are physiologically completely reabsorbed since the specific carriers of these substances are not saturated. When such a substance is administered in acute, its urinary excretion appears as soon as the plasma concentration reaches a critical value which defines the renal threshold of this substance. Above this threshold, the transporters are gradually saturated and the urinary excretion of the substance increases parallel to the filtered charge, making it possible to calculate the maximum reabsorption capacity or Tm of the substance.
For some solutes however, tubular reabsorption is more complex and the tubular reabsorption capacity can not be assessed by a measured or estimated Tm. The most representative example is probably that of calcium, the reabsorption of which is gradually inhibited with the elevation of calcemia. The study of the relationship between plasma concentration and urinary excretion makes it possible to compare the urinary excretion of calcium with that obtained in healthy subjects with identical calcium and filtered charge and to document a possible modification of the tubular reabsorption of calcium.
Exploration of proximal tubular functions:
A number of constituents of the glomerular filtrate are almost completely reabsorbed (glucose and amino acids) or predominantly (phosphates, 70%, bicarbonate, 85%) in the proximal tubule. A decrease in their reabsorption capacity in the proximal tubule is accompanied by abnormally high urinary excretion without variation in plasma concentration (glucose, amino acid) or decreased plasma concentration (phosphorus, bicarbonate) with excretion urinary retention and therefore inappropriate.
An overall impairment of all proximal tubular reabsorption defines renal Fanconi syndrome, among which the numerous medicinal causes are noted.
Tubular reabsorption of phosphate:
The plasma phosphate concentration in the normal subject (close to 1 mmol / l) is above the actual plasma threshold.There is thus a notable physiological urinary excretion of phosphates which accounts for about 30% of the filtered phosphates. The Tm value of DFG (TmPi / DFG) measures the maximum reabsorption capacity of phosphates independently of changes in the filtered charge resulting either from a change in phosphate levels or from a variation in GFR. Its value is in the normal subjects between 0.77 and 1.45 mmol / l of glomerular filtrate.
Fractional excretion and fractional reabsorption of phosphate (known as TRP) should no longer be interpreted directly because they are influenced by the filtered charge and hence by the plasma phosphate concentration and GFR. Thus, when the phosphate levels decrease, the fractional excretion decreases and the fractional reabsorption increases, and vice versa. However, their calculation from plasma and urine concentrations of phosphate and creatinine in a fasting subject makes it possible to derive TmPi / DFG from a nomogram.
The primary decrease in TmPi / DFG is generally associated with a decrease in phosphate levels and should not be confused with the decrease in normal phosphatase TmPi / DFG observed in response to high phosphate inputs or during nephron reduction. In these situations, the decrease in TmPi / DFG represents an adaptation to the increase in intakes or to the filtered phosphate load increased by residual nephron, and should not be interpreted as a primary abnormality of tubular renal tubular reabsorption .
Tubular reabsorption of glucose:
Since the normal plasma glucose value is well below its renal threshold, the normal subject does not eliminate glucose in the urine. Thus, the presence of fasting glycosuria present in normal blood sugar levels demonstrates renal glycosuria. Glucosuria in a 24-hour urine collection should ensure that the subject does not have a carbohydrate intolerance by simply performing an oral glucose test to verify that during this test plasma glucose does not does not rise well above the threshold, which can then explain the glycosuria. Renal glycosuria may remain isolated with no consequence to the patient; it can also integrate into Fanconi syndrome.
Finally, glucosuria preceding the onset of diabetes has been described in MODY III type diabetes in relation to a mutation of the HNF1a gene. The quantification of this renal glucose loss then requires the measurement of glucose Tm / DFG during hypertonic glucose perfusion. The normal value of the glucose renal threshold was 1.4 to 2 g per liter (8 to 11 mmol / l) and that of glucose Tm / DFG of 3 g / l (16 mmol / l).
Tubular reabsorption of uric acid:
Uric acid is the final product of the degradation of free purines. Its daily production is between 600 and 800 mg, eliminated for two thirds in the urine, and one third in the bile, stomach and mainly the intestine, where it is degraded by the uricase of the colonic bacteria. The urinary clearance of uric acid is 9 ± 3 ml / min, ie one tenth of the creatinine clearance.
The presence of hypo-uricemia is due to two physiopathological mechanisms which are distinct but not exclusive: the decrease in the formation of uric acid due to a primary or secondary defect in the xanthine oxidase activity; increased renal clearance of uric acid.
In the absence of apparent saturation of the reabsorption of uric acid, its alteration is evaluated using fractional excretion. These two pathophysiological frameworks can be distinguished by the measurement of uricuria which is collapsed in the first case, preserved in the second with fractional excretion greater than 10%.
Since the urate transport route is common to several organic anions, many generally exogenous substances can compete with uric acid at its transport sites (such as salicylates at low doses) – or hyperuricemic.
After glomerular filtration, the urate undergoes complex bidirectional transport, essentially in the proximal tubule where it is the site of both reabsorption and secretion. The proximal portion of the proximal tubule (S1) is the site of clear reabsorption; the second portion (S2) is the site of a net flow of secretion, followed by a low net reabsorption in the more distal segments (so-called “secretory reabsorption”). Pharmacological tests have made it possible to define “presecretory”, “secretory” and “secretory” modifications. They consist in analyzing the uricosuric responses induced by the combination of acrid administration of pyrazinamide (supposed to selectively inhibit the secretion of uric acid) and benzbromarone (marketed in France until 2003 under the name Desuric ® , which would preferentially inhibit “postsecretory” reabsorption).
Tubular reabsorption of bicarbonate:
In the normal subject, 85% of the filtered bicarbonate is reabsorbed into the proximal tubule. The actual plasma threshold of bicarbonate is slightly higher than the normal concentration of plasma bicarbonate, so urinary excretion is physiologically negligible. In the case of proximal tubular acidosis, decreased bicarbonate reabsorption capacity decreases the plasma concentration until the filtered load drop is sufficient to offset the decrease in tubular reabsorption. The urinary loss of bicarbonate then stops and a stable state is established, characterized by deep metabolic acidosis but a balanced acid balance. Indeed, the net acid excretion is equal to the daily production of fixed acids, since the distal function of acidification of the urine is intact. Urinary pH may theoretically be acidic. However, patients are often explored while their acidosis is partially corrected by an exogenous supply of bicarbonate. This treatment, which artificially maintains its plasma bicarbonate concentration above its reabsorption threshold, explains that the urinary pH observed is frequently greater than or equal to 7, which can lead to an abusive conclusion to distal tubular acidosis. The diagnosis of proximal tubular acidosis is based on the measurement of Tm of bicarbonate in a standardized filtered load situation. The test consists in correcting the plasma concentration of bicarbonate (value greater than 24 mmol / l), usually with hypertonic NaHCO 3 perfusion. A fractional excretion of bicarbonate greater than 15% of a normal bicarbonate (24-26 mmol / l) or better a TmHCO 3- / DFG less than
20 mmol / l of glomerular filtrate can be used to confirm proximal tubular acidosis. It should be noted that in chronic renal insufficiency, the decrease in bicarbonate reabsorption capacity retains all its specific value of proximal tubular involvement because, apart from this situation, renal insufficiency is accompanied by an increase in TmHCO 3- / DFG, a reflection of the reabsorption increased by residual nephron.
Tubular reabsorption of amino acids:
In the normal subject, the plasma concentration of amino acids is also below their reabsorption threshold.
More than 98% of the filtered amino acids are reabsorbed into the proximal tubule and the physiological amino-aciduria is very low.
A pathological amino aciduria is readily detected with urine chromatography. A pathological and non-selective amino aciduria always involves an abnormality of proximal tubular reabsorption. In the presence of a selective aminoaciduria of dibasic amino acids (cystine for example), neutral (methionine, tryptophan, histidine) or diacids (aspartic acid, glutamic acid, and aminoglycinuria), it is absolutely necessary to check the absence of elevation in the plasma of the corresponding amino acids. This measure makes it possible to distinguish a prerenal aminoaciduria by increasing the circulating concentration and the filtered amount of a tubular specific deficit proximal to the transport of a group of amino acids.
Tests for localization of tubular defects of sodium chloride reabsorption:
When the clinical and biological context is strongly compatible with Bartter or Gitelman syndrome, it is possible to better document the NaCl decrease in the dilution segment (which includes Henle’s broad ascending limb and tubule distal distal). Tests may include natriuretic response to diuretics (natriuretic response to decreased furosemide in Bartter’s syndrome, decreased natriuretic response to thiazide diuretics in Gitelman’s syndrome), or indirectly to estimate NaCl re-absorption in the dilution. The latter test consists of measuring free water clearance and chlorine clearance during hypotonic NaCl perfusion. The principle of the test is that the reabsorption of NaCl in the dilution segment makes it possible to subtract osmoles from the tubular fluid and to excrete, in the case of aqueous charge, a large volume of water without osmole, which volume can be quantified by the calculation of the free water clearance (CH 2 O). The NaCl flow delivered to the dilution segment is indirectly estimated by the sum of the chlorine clearance (reflection of the quantity of NaCl delivered, not reabsorbed and excreted in the urine) and the clearance of the free water NaCl delivered to the dilution segment and reabsorbed at this site). The CH 2 O / (CH 2 O + CCl) ratio is used to estimate the fractional re-absorption of NaCl in the dilution segment. This relationship is strongly affected in the course of Bartter’s syndrome and, to a lesser degree, in Gitelman’s syndrome. The clearance of lithium, proposed as a reflection of the reabsorption of NaCl in the proximal tubule, is not very specific in practice.
Exploration of distal tubular functions:
Study of the power of acidification of urine:
Acid loading test:
This test is unnecessary and harmful in a patient with spontaneous metabolic acidosis. On the other hand, in the partial abnormalities of acidification of the urine, the defect of excretion can be compensated by the mobilization of the bone pads. In this case, the pathology can be revealed by bone demineralization or by kidney stones. Biologically, there is no acidosis but hypocitraturia which testifies to the intracellular acidosis associated with hypercalciuria.
An acidifying abnormality in the absence of spontaneous metabolic acidosis (partial distal acidosis) can be investigated by an acid loading test, which consists in administering 2 mmol / kg of NH 4 Cl in aqueous solution. The response is judged within 6 to 8 hours following two criteria: urinary acidification (urine pH should drop below 5.5) and acid excretion rate, which should increase.
Characterization of the defect of acidification of the urine:
The acidification of the urine requires the coupled operation of the main and intercalary cells of the collecting duct.According to the classical scheme, the main cells’ electrogenic reabsorption of sodium is indirectly linked to the generation of a negative potential difference in light that stimulates paracellular Cl reabsorption (which would tend to dissipate the transepithelial potential difference) , secretion of potassium by the main cell and protons by the apical proton pump of the intercalated cells (H + ATPase). Urine acidification creates a pH gradient between the urine and the interstitium favorable to NH 3 diffusion which is trapped and excreted in the urine as NH 4+ .
Secretory defect:
In the secretory defect, the acid secretion by the intercalated cells is initially altered. These defects can be acquired or secondary to gene inactivation of a transporter involved in distal acid secretion. There are two forms of hereditary distal tubular acidosis. The autosomal dominant form is linked to heterozygous mutations in the gene that encodes the Cl / HCO 3 AE1 interchange. Autosomal recessive forms are linked in 80% of cases to homozygous or composite heterozygous mutations of the gene that encodes the B1 and A4 subunits of H + -ATPase.
Defect of voltage:
In the voltage defect, the acid secretion by the intercalated cells is not initially altered but insufficiently stimulated, because the sodium reabsorption is inhibited (example: taking amiloride) or not stimulated (example: hypoaldosteronism). This defect is corrected by maneuvers that increase the difference in transepithelial potential negative light (bicarbonate load, furosemide test). It is generally associated with a hyperkalaemic tendency which inhibits the production and interstitial accumulation of NH 3 / NH 4+ . The low availability of interstitial buffers in hyperkalaemic distal acidosis causes the urinary pH to mislead in an appropriate manner in response to acid loading, while acid excretion is not appropriately stimulated.
A sub-type of voltage defect is the “chloride shunt” described in Gordon’s syndrome, also improperly called pseudohypoaldosteronism type 2, characterized by primary renal NaCl retention with secondary renin-angiotensin retention. It is now known that this acidosis is related to hypoaldosteronism, associated with stimulation of NaCl reabsorption in the distal circumferential tubule which decreases the NaCl load delivered to the collecting channel. The primary abnormality is related to the deficiency of a thiazide-sensitive NaCl cotransporter regulatory protein of the distal tube. The particularity of this syndrome is to be extremely sensitive to thiazide diuretics whose low doses correct hypertension, acidosis and hyperkalaemia.
Gradient defect:
In the defect gradient, acid secretion by the intercalated cells is retained, but there is a tubular inability to maintain the pH gradient. The physiopathological model of this type of defect is tubular acidosis induced by amphotericin B.
Dynamic tests:
These defects are characterized by dynamic tests.
Charge in bicarbonate. It makes it possible to test the acid secretion by measuring the elevation of the pressure of urinary carbon dioxide (PCO 2 ) with respect to the plasma PCO 2 , a reflection of the PCO 2 of the cortical interstitial fluid. The principle of this test is to use urine bicarbonate as a reagent that generates CO 2 in proportion to the rate of acid secretion.
The test can only be interpreted if there is no advanced interstitial nephropathy (evidenced by a sustained urine concentration capacity) and if the bicarbonate is sufficiently high during exploration. Inadequate elevation of urinary PCO 2 (PCO 2 urinary – plasma PCO 2 less than 20 mmHg) indicates a primary defect of acid secretion.
Furosemide test. It consists in stimulating the distal reabsorption of NaCl by the combined administration of mineralocorticoid and furosemide which causes in the healthy subject an acidification of the urines and a stimulation of the acid excretion.
An abnormal response indicates a primary defect of acid secretion.
Relationship between ammoniuria and urinary pH. Finally, it is possible to detect distal tubular acidosis by low interstitial availability of NH 3 / NH 4 + .
Study of the power of concentration / dilution of the urine:
Study of the power of concentration of the urine: tests of water restriction and administration of dDAVP:
Primary impairment of the ability to concentrate urine results in intracellular dehydration usually limited by stimulation of thirst. The earliest sign of a defect in urine concentration is therefore usually a polyuria, defined by a volume of urine greater than 3 l / 24 hours.
To test urine concentration, urinary osmolarity should be interpreted during periods of hypernatremia, whether spontaneous or induced by a water restriction test. The usual water restriction test lasts from 12 to 16 hours and begins the evening before, except in cases of major polyuria greater than 6 l / 24 hours where permanent medical supervision is required. The water restriction is continued until the plasma osmolarity exceeds 290 mOsm / kg water (in the absence of osmotic hole) and the urinary osmolarity stabilizes on three successive urinary collections of 1 hour each. In a healthy non-polyuric subject, urinary osmolarity rises above 850 mOsm / kg of water and urine output is less than 0.5 ml / min. The balance of osmoles, in particular of sodium, is not modified; the moderate weight loss corresponds to the loss of water, about 500 g. At the end of the test (or immediately in a spontaneously hypernatremic patient), administration of nasal AVDD is useful for testing the neurogenic origin (defect of antidiuretic hormone (ADH)) or nephrogenic (lack of concentration of the urine secondary to the absence of a renal response to the hormone).
In their complete forms, it is easy to differentiate nephrogenic diabetes insipidus from neurogenic diabetes insipidus.
In patients whose polyuria is secondary to primary polydipsia, the urine becomes frankly hypertonic after the only controlled hydric restriction. In patients with complete diabetes insipidus, the urinary osmolarity does not exceed 200 to 300 mOsm / kg of water. Administration of AVP results in a sharp reduction in urine flow and a very significant increase in urinary osmolarity in neurogenic diabetes insipidus, but does not significantly elevate urinary osmolarity in nephrogenic diabetes insipidus.
In the more common, moderate tables, the etiologic diagnosis of polyuria is often much more difficult since the subjects then retain a certain ability to concentrate urine in water restriction and may respond partially to the administration of dDAVP. It is then difficult to know whether the primary abnormality in question is a partial defect of ADH secretion or a partial resistance to the renal effect of the hormone (diabetes insipidus neurogenic or partial nephrogenic). It is also sometimes difficult to exclude primary polydipsia, even when the maximum osmolarity in water restriction or after dDAVP stabilizes at a value of less than 850 mOsm / kg of water. The interpretation must then take into account the specific effect of chronic polyuria, which considerably alters the power of concentration of the urine. It is therefore understandable how difficult it is to differentiate polyuria from polyuria in a polyuria linked to nephrogenic or partial neurogenic diabetes insipidus.
Interest in the dosage of the antidiuretic hormone and the assessment of thirst:
The sensation of thirst can be measured using a self-assessment scale. The circulating concentration of DHA is measured by radioimmunoassay, under conditions which make it possible to limit its degradation. In situations of partial diabetes insipidus, it is particularly important to measure the circulating concentration of ADH: the maximum urinary osmolarity is obtained at the price of a very high ADH secretion in the case of nephrogenic diabetes insipidus or on the contrary low in neurogenic diabetes insipidus which associate with peripheral sensitization to the hormone. It is sometimes useful to characterize the response of ADH secretion and thirst in response to hyperosmolarity induced by hypertonic NaCl perfusion. In a patient with nephrogenic diabetes insipidus or primary polydipsia, the plasma ADH concentration normally rises while the ADH values remain low and inappropriate in a subject with neurogenic diabetes insipidus.
Study of the dilution power by the aqueous charge test:
The body has an impressive water removal capacity, since the kidneys are theoretically capable of excreting nearly 20% of the GFR, bringing the theoretical water excretion capacity to more than 20 l per 24 hours as evidenced by Extremely high diuresis observed exceptionally in clinical pathology.
This capacity of water excretion depends on three elements: the DFG, the dilution capacity and the osmotic inputs of the subject, all of which must be evaluated in parallel. Renal insufficiency alone can not explain the onset of hyponatremia unless it is severe (DFG less than 20 ml / min).
The dilution capacity is appreciated in a subject whose plasma osmolarity is less than or equal to 280 mOsm / kg of water, which results in an increase in diuresis and decrease in urinary osmolarity to a minimum value in a healthy subject less than or equal to 100 mOsm / kg of water, which defines the intrinsic dilution capacity of the urine of the subject.
A urinary osmolarity of more than 100 mOsm / kg of water (150 in the elderly), which is contemporary with hypotonic hyponatraemia, indicates an alteration in the dilution capacity. In the absence of spontaneous hyponatremia, the dilution state may be induced by an aqueous charge test. Oral administration of an amount of water equivalent to 20 ml / kg of body weight within half an hour causes an expected decrease in plasma osmolarity of the order of 10 mOsm / water and serum sodium of the order of 5 mmol / l. This test allows us to evaluate the renal capacity of water excretion, which normally allows to eliminate 70% of the water load in less than 4 hours.
The lack of dilution is related to the stimulation of the secretion of ADH by a true or effective hypovolemia (volumic stimulus of ADH), or related to a table of SIADH.
Osmotic intakes are evaluated by the amount of osmoles excreted in a 24-hour urine collection. Low osmotic intake limits the ability of water excretion and is of particular importance in subjects with a small alteration in dilution capacity.Thus, an elderly person with a slight (physiological) alteration in his dilution capacity (minimum urinary osmolarity 150 mOsm / kg of water) has a maximum diuresis of 6 liters if his osmotic intake is 900 mmoles per 24 (900/150 = 6) but can excrete only 2 liters of water in the urine if its osmotic intake is low, 300 mmol / 24 hours (300/150 = 2). This person then has a propensity to develop hyponatremia, provided that his diet is essentially based on liquid intakes (soups, tea, herbal teas, etc.). This table is described as tea and toast syndrome.
Genetic diagnosis of hereditary tubulopathies:
A large number of hereditary tubulopathies are maintained lucid at the molecular level. Information about these orphan diseases, their clinical presentations, and the care or diagnostic centers specialized in these diseases can be consulted on the ORPHANET website.