 Introduction:
Introduction:
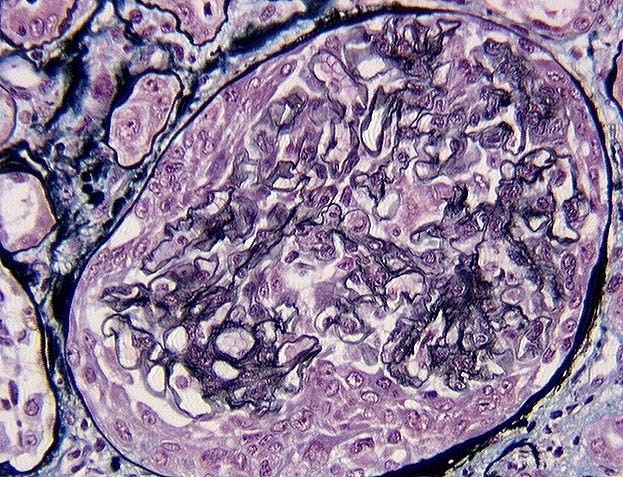
Extracapillary glomerulonephritis (GNEC) is characterized by the presence of a more or less circumferential crescent cell proliferation surrounding the glomerular flocculus and the so-called endocapillary cells (endothelial and mesangial).
There is some confusion with the clinical notion of rapidly progressive glomerulonephritis (GNRP), which refers to rapid renal failure in a few weeks in a biological context of glomerular disease. Besides the fact that a term of pathological anatomy can not be synonymous with a clinical fact, these two entities are not strictly superimposable in human pathology. Indeed, some GNECs can lead to slow degradation of the renal function, and conversely other histological damage (hemolytic uremic syndrome, scleroderma …) sometimes reveal a GNRP table.
All the diseases that can induce GNECs have been progressively dismembered into several groups corresponding to the beginning of etiopathogenic classification. The disease of glomerular basal antimembrane antibodies (MBG) was the first and remains the only clearly identified type. In the second inhomogeneous group of GNECs with generally granulous deposits of immunoglobulins (Ig), the formation of immune complexes has been incriminated, but their pathogenic role has not always been demonstrated. More recently, the third group of GNECs without significant Ig deposition was characterized most often by the presence of cytoplasmic cytoplasmic anticonvulsant circulating antibodies of neutrophilic polynuclear cells (ACPN) with or without systemic vasculitis lesions. Moreover, some primitive glomerulopathies sometimes become complicated by extracapillary proliferation at a more or less late stage of their evolution.
In this chapter, we develop more fully the pathologies not treated in other parts of the book: vasculitis with ACPN and Goodpasture’s disease.
Pathological anatomy:
DESCRIPTION OF INJURIES:
The diagnosis of GNEC is an anatomopathological diagnosis defined by a characteristic aspect in optical microscopy.
Immunofluorescence examination provides essential elements for the etiological diagnosis. This lesion is not specific but reflects a particularly severe attack of the basal membrane.
Definition:
The very peculiar aspect of the glomerular lesions, which gave to these glomerulopathies the imaged term of crescent glomerulonephritis, was described by Volhard in 1914. The crescent is an elementary lesion defined by the accumulation of inflammatory elements (monocytes macrophages, polynuclear, lymphocytes and sometimes giant cells) and above all of epithelial cells mixed with fibrin, in the space of Bowman.
Morphological aspects of croissants:
Croissants are presented in very varied histological aspects according to the importance of the epithelial cell proliferation and of the inflammatory infiltrate, and according to the evolutionary stage. At an early stage, it is essentially constituted by a fibrinous network enclosing a few inflammatory cells and especially epithelial cells. The latter have a moderately abundant and pale cytoplasm, and are grouped into clusters producing weakly proliferative segmental lesions. Then, epithelial proliferation is accentuated: the cells are arranged in concentric strata and repel partly or totally the flocculus. If the Bowman capsule is intact, the epithelial cells remain largely predominant. On the other hand, if the Bowman capsule is broken, the inflammatory elements become predominant. At a later stage, the cells appear less numerous and enclosed in a collagenous network: the crescent is called “fibrocellular”. Gradually, the cells become scarce to disappear, leaving room for a dense collagen blade: the crescent is called “fibrous”.
Morphological aspects of the underlying flocculus:
Its appearance, highly variable, depends on the intensity and the etiology of the GNEC. It may be normal, have occasional ruptures of the capillary basement membrane, or wide ranges of segmental necrosis. It may be the site of endocapillary proliferation and include endangular or extramembrane mesangial immune deposits. Finally, it can be almost completely destroyed, persisting only in the form of a few flaps of basal membranes, or totally sclerotic as “sealing bread”.
Other related lesions:
The interstitium is most often oedematous, the site of a predominantly mononuclear polymorphic infiltrate whose perglomerular and perivascular reinforcement evokes vasculitis. The tubular lumens are frequently congested with hematic cylinders and the tubular epithelium shows signs of generally minor suffering. The vessels appear mostly normal, but careful examination of serial sections is essential for the detection of lesions of necrotizing and granulomatous angiitis.
Diagnostic criteria for extracapillary glomerulonephritis:
The histological definition of GNECs is not unanimous. The debate focuses on the percentage of glomeruli affected by proliferation and on the volume of croissants. According to the authors, 30 to 80% of affected glomeruli are required to carry the diagnosis. But should this percentage take into account the sclerotic glomeruli? At present, the French authors retain the number of 50% of glomerules with crescents formed from at least two cell strata and occupying more than 50% of the surface of the filtration chamber. However, Cameron stresses the fragility of such a definition since, according to the cutting plane of the same glomerulus, the latter may appear normal, or the crescent, if it appears, may be segmental or circumferential. To be representative, this percentage must therefore be evaluated on a minimum number of glomeruli, estimated at 20 for Cameron, for most authors and requires that a biopsy be studied on serial sections.
Immunofluorescence:
Common immunofluorescence techniques provide the clinician with two types of information:
– on the one hand, the detection of fibrin deposits within the crescent characterizes acute lesions without prejudging the etiology of the disease;
– on the other hand, the appearance of Ig deposits and complement fractions allows the classification of GNECs into three distinct groups that correspond to different immunopathological mechanisms:
Group I gathers the Ig-linear GNECs along the basal membrane;
Group II, which is much more heterogeneous, groups together GNECs with granular deposits of Ig;
Group III is that of the GNECs with little or no deposit of Ig, also referred to by the term pauci-immune glomerulonephritis, which is in fact inadequate.
ETIOPATHOGENIA:
Triggers and Amplifiers:
Real perforations of MBG were observed by electron microscopy. These defects of the basal membrane would play a pathogenic role because they cluster at the level of thrombosed capillary loops and are generally associated with necrosis of endothelial cells and podocytes. The size of these breccias is sufficient to allow the passage of serum proteins and cellular elements. These breccias are secondary to the action of proteolytic enzymes and oxygenated radicals released by leukocytes in contact with the glomerular endothelium, activated by various factors (complement fractions, Ig Fc fragment, T lymphocytes or autoantibodies).
Tissue factors of glomerular origin initiate the activation of coagulation, which results in the formation of thrombin and fibrin, which accumulates in the urinary chamber. However, experiments in fibrinogen-deficient mice have shown that fibrin formation is not an essential step in crescent formation. In experimental models, plasminogen and plasminogen activators limit the consequences of coagulation activation. Furthermore, tissue factors induce an increase in the expression of class II human leukocyte antigen (HLA) molecules by the glomerular resident cells, which are essential for the development of experimental extracapillary glomerulonephritis. Apart from its procoagulant action, thrombin could promote inflammation and crescent formation by activating the receptor for thrombin PAR-1. Activated macrophages participate in this procoagulant activity by producing tissue thromboplastin, and also indirectly through increased production of interleukin (IL) 1 and tumor necrosis factor (TNF) -α which depress the expression of anticoagulant activity by endothelial cells (heparin-like proteoglycans, thrombomodulin, prostacyclin).
Proinflammatory cytokines, in particular IL1 and TNF- a , in dependence on the nuclear transcription factor NF-kappa B, increase the expression of adhesion molecules (selectin, integrin) on the surface of endothelial cells and promote activation and diapedesis of leukocytes. In animals, the administration of the IL1 receptor antagonist (IL1-RA), the soluble TNF-α receptor (sTNFr55), or anti-leukocyte function associated antigen (LFA) -1 or anti -intercellular adhesion molecule (ICAM) -1, prevents the formation of croissants in some experimental models.
The migration of inflammatory cells between the endothelial cells to the urinary chamber takes place according to a gradient of chemotactic agents: complement fraction (C5a), chemokines (monocyte chemoattractant protein [MCP] -1, IL8, RANTES …), lipids leukotriene LTB4, platelet activating factor [PAF], etc.), extracellular matrix proteins (osteopontin), etc. These molecules form a particularly redundant system and consequently difficult to control by therapeutic agents.
However, recruitment and activation of macrophages is a key step in crescent formation, regulated primarily by certain macrophage migration inhibitory factor (MIF) macrophage inflammatory protein (MCP-1). Moreover, the extracellular matrix facilitates the migration and interaction of infiltrating cells with growth factors, but also promotes the proliferation of glomerular epithelial cells.
Metalloproteinases (MT1-MMP and MMP-2) participate in alterations of the extracellular matrix. Moreover, podocytes are involved in the early formation of bridges between MBG and the proliferating parietal epithelium.
Cellular immunity plays a decisive role. CD4 and CD8 deficient mice demonstrated the predominant role of CD4 + .The models of murine extracapillary glomerulonephritis are mainly dependent on the Th1 component. Indeed, the lesions are aggravated by the IL12 produced by the mesangial cells, which promotes the Th1 response, and by the gamma interferon (IFN) produced by the Th1 lymphocytes, which participates in the macrophage activation. The lesions, on the other hand, are improved by the administration of IL4 or IL10 which inhibits the Th1 response. IL4-deficient mice with an amplified Th1 response develop more severe GNECs. However, CD8 + lymphocytes also play a role in some experimental models.
Other regulatory elements have been described, including receptors for the Fc fraction of Ig (FcR), inducible NO synthase (iNOS), oxygenated radicals, prostaglandins, and transforming growth factor (TGF) -b which stops the inflammatory process at the cost of fibrosis.
Identification of crescent cells:
Immunohistochemical techniques using monoclonal antibodies allowed the cells responsible for epithelial proliferation to be identified as essentially parietal epithelial cells. Thrombin and certain growth factors, secreted by macrophages, among others, were incriminated.
Inflammatory elements of the crescent of human GNECs essentially gather macrophages and T lymphocytes. The proportion of macrophages increases with rupture of the capsule of Bowman and testifies to severe lesions and worse prognosis. T lymphocyte analysis of flocculus and crescent, and interstitial and peri-glomerular infiltrate revealed a predominance of CD4 + and the presence of activated cells (RIL2 positive). Peripheral lymphocytes and macrophages could be responsible for rupture of the Bowman capsule.
Fibrous evolution:
The extinction of the inflammatory process is marked by the accumulation of extracellular matrix and the evolution towards fibrosis of the crescent. TGF- b produced by glomerular cells would be the main regulating element of this process. The administration of anti-TGF-β antibodies, or of a proteoglycan, the decorin, which neutralizes all the isoforms of this cytokine, inhibits the fibrosis of the croissants in experimental models. Angiotensin II is a potent stimulant of TGF- b production and inhibition of the renin-angiotensin system may limit the processes of glomerular fibrosis in experimental models. The nature of collagen has been identified. It would be more of glomerular type (laminin and collagen IV) as long as the capsule remains intact, and rather of the interstitial type (type III) when the capsule is broken.
A better knowledge of the pathophysiology of extracapillary proliferation will soon make it possible to propose particularly effective treatments, capable of acting at different stages of the disease and the evolution of the crescent.
Main etiopathogenic factors:
GLOMERULONÉPHRITES WITH GLOBULAR BASAL ANTIMEMBRANE ANTIBODIES:
The pathogenicity of the anti-MBG antibodies has been demonstrated by animal disease transfer experiments. These antibodies are most often IgG and exceptionally IgA or IgM. The level of antibodies is generally fairly well correlated with the activity of the disease and makes it possible to adapt the therapy.
Anti-MBG antibodies are directed against a structure of type IV collagen. Collagen is a fibrillar structure corresponding to the assembly of three chains. Each chain is composed of a so-called “collagenic” portion characterized by the presence of glycine every three amino acids, allowing the coiling of these triple helix chains, and a carboxyterminal globular portion called the “non-collagenic” domain.
Six non-collagenous domain variants have been identified for type IV collagen: alpha-1 and 2 are coded by chromosome 13, alpha-3 and 4 by chromosome 2, and alpha-5 and 6 by chromosome X. L epitope recognized by the anti-MBG antibodies is contained in the non-collagenous NC1 domain of the alpha-3 chain of collagen type IV (encoded by the COL4A3 gene). The alpha-3 chain of collagen type IV is present mainly in the basal glomerular and alveolar pulmonary membranes. Alport’s syndrome, in its classical juvenile X-linked form, is characterized by an abnormality of the COL4A5 gene, which prevents normal synthesis not only of alpha-5 chains but also of alpha-3 and 4. This explains why patients with ALP syndrome may develop anti-MBG antibodies directed against the alpha-3, 4 and 5 chains of collagen IV after transplantation. However, the clinical expression of the disease probably depends on the ability to develop an immune cell response. The anti-MBG antibodies have a high affinity for their antigen, causing prolonged aggression of MBG despite attempts to extract by plasma exchange. The immune complex formed in situ results in an inflammatory process secondary to adhesion and activation of leukocytes by adhesion molecules (integrins rather than selectins) and Fc gamma receptors, rather than by complementary activation.
Apart from genetic factors (HLA DR), the disease appears to be determined by environmental factors (toxic or mechanical by lithotripsy). Infections can aggravate the disease via pro-inflammatory cytokines. Smoking encourages the development of pulmonary haemorrhages. There may be a dissociation between pulmonary and renal involvement, probably because the pulmonary capillary is not fenestrated and therefore the basal membrane is protected by the vascular endothelium.
GLOMERULONÉPHRITES WITH GENERALLY GRANULAR DEPOSITS OF IMMUNOGLOBULINES:
Infectious diseases:
Certain bacterial infectious agents can induce GNEC associated with immune complex deposition, generally causing complement activation by the conventional route. The localization of the deposits (subendothelial or subepithelial), the germ and the origin of the infection in question may explain the different histological presentations, as well as the clinical signs and prognosis of these post-infectious renal lesions. The most common microorganisms involved are beta-haemolytic streptococcus A and epidermidis and golden staphylococci, particularly in shunt nephritis and acute endocarditis. The glomerular lesion is a diffuse proliferative and exudative GN, associated with extracapillary proliferation either directly or indirectly.
Certain germs, in particular Staphylococcus epidermidis, are capable of inducing direct lesion of the glomerular capillary without Ig fixation, and the serum levels of the C3 and C4 fractions of the complement sometimes remain normal. Genetic and environmental factors seem necessary for the induction of the disease, because in an epidemic to one of these “nephritogenic” germs all patients will not have glomerulonephritis and the clinical presentation will be different from a patient to the other. Other bacterial infections are exceptionally complicated by GNEC.
Bacterial infections are more rarely associated with glomerulonephritis without deposition of idiopathic immune complexes, or with systemic vasculitis with ACPN.
Viral diseases are rarely complicated by GNEC. Hepatitis B may be associated with extramembraneous glomerulonephritis, but extracellular proliferation remains exceptional. Hepatitis C is sometimes complicated by mixed cryoglobulinemia type 2 with membranoproliferative glomerulonephritis type 1, but extracapillary proliferation remains exceptional. Periarteritis nodosa sometimes develops during hepatitis B or C, but these vasculitides affecting the middle trunks cause glomerular ischemia without GNEC. GNEC is exceptional in a cytomegalovirus (CMV) infection, or acquired immunodeficiency syndrome (AIDS).
Systemic lupus erythematosus:
Extracapillary proliferation occurs in focal (Class III) and especially diffuse (Class IV) proliferative GNs, secondary to immune complex deposits. Diagnosis is made in front of massive subendothelial and endomembranous mesangial deposits of IgG, A and M, C3, C4 and Clq, with the presence of hematoxic bodies. More rarely, it is a hemolytic uremic syndrome with or without antiphospholipid antibodies or exceptional lupus vasculitis.
Rheumatoid purpura:
A diffuse extracapillary proliferation can reach up to 100% of the glomeruli during rheumatoid purpura. Initial renal involvement is often more severe in adults, but the renal prognosis at a distance remains comparable. It may occur during pregnancy, may be associated with liver cirrhosis and may recur in post-breast. There are borderline forms with vasculitides with ACPN of isotype IgA or IgG.
GLOMERULONÉPHRITES WITH CYTOPLASMIC ANTIBODIES OF NEUTROPHILIC POLYNUCLEARS:
Three vasculitides are usually associated with the presence of ACPN: Wegener’s granulomatosis, microscopic polyangiitis, and Churg and Strauss’s disease. Of these, only the first two are frequently associated with GNRPs.
The situation is similar in children.
ACPNs are generally IgG but sometimes also IgM in the acute phase of the disease. These antibodies are directed against proteins contained in granules of neutrophilic polynuclear cells. The two main antigenic specificities recognized by ACPN in systemic vasculitides are: proteinase 3 (PR3) and myeloperoxidase (MPO). The anti-PR3 antibodies produce a granular cytoplasmic fluorescence on alcohol-fixed neutrophils (C-ACPN), and anti-MPOs a perinuclear fluorescence (P-ACPN) due to the redistribution of their antigenic target around the nucleus after fixation of polymorphonuclear drugs. There is no perfect correlation between antigenic specificity and clinical expression of the disease.
However, most patients with Wegener’s granulomatosis have anti-PR3 antibodies, while a larger proportion of patients with microscopic polyangitis or “idiopathic” GNEC have anti-MPO antibodies.
The arguments in favor of the pathogenicity of ACPN remain indirect in the absence of a study of transfer of the ACPN in the animal or of demonstrated motherofoetal transmission. The specificity of ACPN for systemic vasculitis approaches 99%. An increase in ACPN rates generally precedes clinical relapse. Patients without detectable ACPN do not normally have a relapse, whereas the persistence of a significant level of ACPN in remission increases the risk of relapse.
On the other hand, experimental work has shown that ANCA can induce stimulation of neutrophils with production of oxygenated radicals and degranulation of proteolytic enzymes, as well as lesions of endothelial cells in culture.Monocytes are also activated by ACPN. These events require priming of neutrophils with inflammatory cytokines that allow the translocation of ACPN target antigens to the surface of the cell. The level of expression of target antigens on the surface of neutrophils may be a risk factor for the disease. Target antigens could also be released from adjacent neutrophils and passively adsorbed to the cell surface. The antigen is bound by the ACPN, and the Fc fraction of the autoantibody binds to the Fc c RIIa and RIIIb receptors, resulting in the transduction of an intracellular activation message, which also requires other cofactors. Neutrophil activation by ACPN requires neutrophil adhesion through integrins, which is facilitated by ACPN. Activation is finally amplified by regulatory loops. Furthermore, ACPN accelerates apoptosis of neutrophils and interfere with the clearance of apoptotic bodies by macrophages.
The cause of the appearance of ACPN is often unknown. During neutrophil apoptosis, the ACPN target antigens are exposed to the surface of the cells. A massive exposure of apoptotic bodies results in a breakdown in tolerance and in the appearance of ACPN. An abnormality of the regulation of neutrophil apoptosis (increase in production, abnormal clearance) could therefore play a role in the genesis of the disease.
However, factors other than ACPN could contribute to the genesis of these diseases. There is certainly a role for cellular immunity in these patients, but the rise of ACPN seems to precede the increase in soluble IL-2R. A defect in the protease / anti-protease balance has been reported in patients with anti-PR3 antibodies and an alpha-1-antitrypsin deficiency, an inhibitor of proteolytic activity of PR3, and a factor in the severity of the disease.
General Clinical Description:
The clinical picture is generally that of a GNRP with a degradation in few weeks of renal function, associated with haematuria sometimes macroscopic and proteinuria rarely nephrotic. High blood pressure is mostly present in the most severe forms. This table should lead to a renal biopsy but also a serological survey with, in particular, research of ACPN which makes it possible to guide the diagnosis quickly. Some patients, however, present an insidious installation of the disease with successive thrusts that can go unnoticed.
Principal diseases responsible:
VASCULARITIES WITH NEUTROPHILIC POLYNUCLEAR ANTIYTOPLASM ANTIBODIES (ACPN):
The vasculitis with ACNP responsible for GNRP is mainly Wegener’s granulomatosis and microscopic polyangiitis.These are necrotizing vasculitis mainly affecting small vessels. Wegener’s granulomatosis is characterized by inflammatory giant cell granuloma of the airways or renal interstitium. However, the diagnosis of Wegener’s disease is often made in the absence of granuloma on certain particularly suggestive clinical signs: chronic nodules, sinusitis or chronic otitis media resistant to antibiotic treatment, crusty rhinitis, nasal sagging by cartilage collapse, retroorbital tumor or subglottic stenosis. Microscopic polyangiitis is a vasculitis without granuloma, asthma or eosinophilia, sometimes limited to the kidney (“pauci-immune” glomerulonephritis). The frequency of these pathologies is increasing. These diseases occur somewhat more often in the middle-aged man and are more rare in children. Some family cases have been described. Their diagnosis is most often evoked before a general syndrome associating fever, myalgia, arthralgia or one of many characteristic extrarenal attacks.
In Wegener’s disease, there is sometimes an old history of crusting rhinitis, epistaxis, even partial destruction of the cartilages of the nose, sinusitis, otitis media with hypoacousia, laryngitis or tracheitis, or even subglottic inflammatory stenosis.
Pulmonary signs are present in 90% of patients at the time of diagnosis with cough, dyspnea, excavated pulmonary infiltrates and sometimes pleurisy on chest radiographs. Hemoptysis and sometimes a dramatic array of alveolar hemorrhage with acute respiratory distress syndrome may be observed. Pseudotumorous otorhinolaryngological or pulmonary presentations are sometimes confused with cancers. Other extrarenal sites are ocular (conjunctivitis, uveitis, retinitis, optic neuritis or retroorbital pseudotumor), neurological (mononeuritis more often than cerebral vasculitis), muscular (myalgia), articular (arthralgia), cutaneous (rash, palpable purpura) (pericarditis, myocarditis, endocarditis), and unusually urological (prostate, ureter), digestive (buccal ulcers, abdominal pain, digestive bleeding).Border forms exist with Goodpasture’s syndrome, Churg and Strauss’s disease, polychondritis and inflammatory colitis.
The circumstances of the discovery of microscopic polyangeitis are rather an alteration of the general state, skin signs (rash, necrotic vascular purpura palpable with histological leucocytoclastic vasculitis), ocular (but not retroorbital), sometimes neurological, or pain muscles and joints. Renal involvement may be at the forefront, or even isolated, sometimes evolving through successive surges, or even in a chronic mode. Other patients have a GNRP with renal failure requiring rapid dialysis, and sometimes a hemorrhagic alveolitis comparable to that of Wegener’s granulomatosis or Goodpasture’s disease.
ACPNs are generally found to be present in systemic active forms. The diagnostic value of ACPN increases if an enzyme-linked immunosorbent assay (Elisa) specific for PR3 is associated with the indirect immunofluorescence, defining the cytoplasmic (C-ACPN) or perinuclear (P-ACPN) of DFO. The antigenic target is more often PR3 than MPO in Wegener’s disease, and inversely more often than MP3 in microscopic polyangitis and pauci-immune glomerulonephritis. Despite a positive predictive value of an ACPN test greater than 99% in a rapidly progressive glomerulonephritis, the diagnosis should generally be confirmed by a renal biopsy before starting a heavy immunosuppressive therapy.
GOODPASTURE DISEASE:
Goodpasture’s disease should be distinguished from Goodpasture’s syndrome, which combines acute hemoptotic respiratory failure with GNRP, and the most frequent aetiology of which is microscopic vasculitis (Wegener and microscopic polyangiitis). Limited forms of the kidney are described.
It occurs a little more often in humans, with two frequency peaks: between 20 and 30 years, and after 50 years. This rare disease sometimes occurs in an epidemic fashion, without a responsible infectious agent being identified. Patients present with rapidly increasing dyspnea and hemoptysis, the importance of which is not correlated with the abundance of alveolar hemorrhage, especially in smokers and acute edema of the lung. The chest radiographs show hilarious opacities predominant to bases and then diffuse. Renal involvement is typically a revealing or secondary-onset hematural GNRP.
The diagnosis is based on the detection of circulating anti-MBG antibodies and along the pulmonary alveolar basal MBG and membranes.
Prognostic criteria:
VASCULARITIES WITH ANTIBODIES ANTICYTOPLASM OF POLYNUCLEARS:
The antigenic specificity recognized by the ACPN influences the evolutionary mode of the disease. Indeed, patients with anti-PR3 antibodies are significantly younger, have a more rapidly progressive renal failure, a disease affecting more different organs, are more readily entering remission during treatment, but are more prone to relapses disease.Patients with anti-MPO antibodies tend to have a chronic insidious disease, but overall, renal survival is not influenced by the antigenic specificity of ACPN at a distance from the initial diagnosis.
At the time of diagnosis, the factors that influence renal survival are: creatinine, proteinuria, degree of arteriolar fibrosis, interstitial fibrosis or a low percentage of healthy glomeruli, but not the percentage of croissants. However, many patients with renal insufficiency requiring dialysis recover independent renal function through treatment. The initial level of ACPN antibodies has no prognostic value, but high levels of anti-MPO antibodies in remission are associated with a risk of end-stage renal disease.
Risk factors for early death include cytoplasmic ACPN, alveolar hemorrhage, higher activity score, histologically documented granuloma, or associated alpha-1-antitrypsin deficiency.
GOODPASTURE DISEASE:
Patients with Goodpasture disease generally have a darker prognosis and renal function recovery at the dialysis stage is exceptional. However, they are less likely to relapse from the disease.
The borderline forms between ACPN vasculitis and Goodpasture’s disease have an intermediate prognosis.
Treatment:
GENERAL RULES:
The symptomatic treatment is that of any acute renal failure and possible extrarenal localizations of a disease of system. Specific treatment is required for infectious or toxic GNECs.
TREATMENT OF A TYPE III EXTRACAPILLAR GLOMERULONEPHRITE WITH POLYNUCLEAR ANTICYTOPLASM ANTIBODIES:
Induction treatment:
The induction protocol for GNRP or threatening extrarenal localization, in particular alveolar hemorrhage, combines cyclophosphamide per os (2 mg / kg / day) or intravenous bolus (15 mg / kg / bolus every 2 to 3 weeks ) and prednisone (1 mg / kg / d). Long-term oral cyclophosphamide protocols 1 year or longer were associated with cumulative doses responsible for serious adverse events: opportunistic infections with Pneumocystis carinii or cytomegalovirus and especially urinary tract cancer sometimes more than 10 years after the end of exposure . After only 1 year of oral cyclophosphamide, the risk of bladder cancer is already multiplied by 11.
In the case of continuous oral treatment, infections with Pneumocystis carinii must be prevented by a co-formulation of cotrimoxasole, or especially pentamidine aerosols, which are less providers of leucopenia. Hematuric cystitis is prevented by the association of mesna.
In an attempt to decrease the cumulative dose of cyclophosphamide, several studies compared the continuous intravenous bolus oral route, but no study had the power to conclude. The oldest studies only recommended monthly boluses and the most recent boluses every 15 days in the first month and then every 3 weeks. A European study is currently under way (Cyclops) to try to answer definitively this question. However, if the disease resists cyclophosphamide boluses every 2 weeks, it is currently recommended to switch to continuous oral treatment. The dose of cyclophosphamide should be decreased in elderly patients or in renal insufficiency.
In case of renal insufficiency requiring dialysis, plasma exchange, or boluses of Solu-Medrol t (1 g / d for 3 days) can be added. The latter two strategies are currently compared in a European study (Mepex). Many other induction treatments have been used, often in refractory forms: immunoadsorption on protein A, 15-deoxyspergualin, leflunomide, intravenous Ig, antilymphocyte serum, anti-TNF, antiintegrin or aplasing bone marrow transplantation chemotherapy.
Maintenance Treatment:
After this phase of induction, the remission is generally obtained and the choice of the maintenance treatment is discussed. Doses of corticosteroids are decreased to a dose of less than 5 to 10 mg / d at 6 months. To decrease the cumulative dose of cyclophosphamide, a relay by azathioprine is possible after the third month without increasing the risk of relapse. It should be borne in mind, however, that azathioprine increases the risk of skin cancer. Because staphylococcal nasal carriage is associated with a risk of relapse of Wegener’s disease, cotrimoxazole has been used successfully in these patients.
However, a study comparing methotrexate to cotrimoxazole demonstrated the superiority of methotrexate for maintaining the remission of Wegener’s generalized forms. Cotrimoxazole should therefore be reserved for the localized oto-rhinolaryngological forms of the disease. Methotrexate may, however, be used as a relay of cyclophosphamide after remission. Mycophenolate mofetil may also be used in this indication. Intravenous Ig can improve some difficult situations, but the benefit of this treatment does not seem to be maintained beyond 3 months.
After discontinuation of immunosuppressive therapy, ACPN should be monitored closely (every 2 months) as a rise in their rate predicts a risk of relapse.
Such an upturn should lead to a further approximation of the biological balance sheets in order to begin an adapted treatment at an early stage, but the value of introducing treatment before clinical relapse remains very controversial.
These patients relapse more often in dialysis than after renal transplantation and immunosuppressive maintenance therapy must sometimes be prolonged. Transplantation is not contraindicated in cases of persistent ACPN positivity.
TREATMENT OF GOODPASTURE DISEASE:
The standard treatment combines oral corticosteroids (1 mg / kg / day), oral cyclophosphamide (2 to 3 mg / kg / day) and daily plasma exchange for 2 weeks or until anti- MBG. At a stage requiring dialysis, recovery of renal function is exceptional.
Protein A immunoabsorption, despite increased IgG subtraction, is not superior to plasma exchange. The cyclophosphamide is prolonged for 2 to 3 months and the corticosteroids are stopped in 6 to 9 months, with a minimal risk of relapse. Transplantation may be considered if autoantibodies have disappeared.
OTHER SELF-IMMUNE EXTRACAPILLARY GLOMERULONEPHRITS:
The treatment of GNECs related to bacterial infectious diseases is based on antibiotic therapy, although corticosteroids and even immunosuppressants are sometimes proposed.
In lupus proliferative glomerulopathies of classes III and IV, the association of cyclophosphamide or mycophenolate mofetil with corticosteroids seems to improve the renal prognosis. Plasma exchange, on the other hand, seems unnecessary.
In rapidly progressive forms of IgA-containing glomerulonephritis with or without rheumatoid purpura, corticoid and immunosuppressive therapy appears to improve progression, although the renal prognosis remains poorer than in ACPN-treated forms, even when the treatment involves plasma exchange.
Conclusion:
A rapid etiologic diagnosis is necessary to avoid the installation of irreversible kidney damage and to allow a suitable treatment.
In this emergency context, certain errors can have dramatic consequences (septic state interpreted as an outbreak of autoimmune disease and treated with immunosuppressants …). The rapidity of the therapeutic intervention and the etiology determine the renal prognosis but also extrarenal and sometimes vital.