 Introduction:
Introduction:
Membranoproliferative glomerulonephritis (GNMP), also called mesangiocapillar, parietoproliferative, lobular or hypocomplementemic glomerulonephritis, constitute a set of glomerulonephritis characterized by mesangial proliferation and variable thickening of the capillary walls.Immunofluorescence and electron microscopy distinguish three types of GNMP, depending on the nature of the deposits and the appearance of the capillary walls.
Immunologically, they often associate with hypocomplementemia and in some cases (GNMP type II) with the presence of a particular autoantibody, the nephritic factor (C3Nef).
Type I GNMPs are generally considered to be diseases associated with immune complex deposition. The secondary forms, by far the most frequent, are dominated by infections, with a special place for the hepatitis C virus (HCV).
Epidemiology:
In the industrialized countries, GNMPs today represent less than 5% of adult glomerulonephritis, whereas they were found in almost 20% of cases, 25 years ago.
Currently, it is assumed that they account for 5% of nephrotic syndrome cases in children and 10% of adult cases.
In developing countries, they are much more common, accounting for as much as 40% of glomerular nephropathies in Mexico, but the frequency of type II GNMPs is superimposed on that observed in Western countries.
The marked decrease in GNMP cases in Europe and North America suggests that these nephropathies had an infectious origin. Measures limiting the transmission of HCV (avoidance of positive subjects for blood donations) probably explain, at least in part, this decline.
The primitive forms of type I GNMP, which are increasingly rare, generally affect subjects between the ages of 5 and 30 years without gender predominance. Type II GNMPs are found in the vast majority of cases in subjects less than 20 years of age.
Classification. Pathological anatomy :
The combination of the various techniques of optical or electronic microscopy and immunofluorescence allowed the subdivision of GNMP into three different types depending on the nature of the thickening of the capillary walls.
– Type I is the most frequent: 70% of GNMP. Interposition of the mesangial tissue produces an apparent splitting of the paro-capillary. This mesangial interposition is, in general, associated with subendothelial deposits. This is the GNMP with subendothelial deposits.
– Type II accounts for about 15% of GNMP. It is characterized by a dense transformation of the basal membrane with or without mesangial interposition: it is the GNMP with dense deposits intramembranous.
– Type III, about 15% of cases, involves interruptions of the basal membrane associated with intramembrane dense deposits.
On repeated biopsies, there was no evidence of a change from one type to another.
MEMBRANOPROLIFERATIVE MEMBRANOPROLIFERATIVE DEFECT WITH SUB-ENDOTHELIAL DEPOSITS: TYPE I
In optical microscopy:
Three main elements characterize type I and are well recognizable if the appropriate colorations, in particular trichrome and silver, are used:
– the proliferation of mesangial cells with an increase in mesangial substance: it is in this type that it is most abundant;
– the mesangial interposition between the lamina densa and the endothelium giving a double contour appearance to the basal membrane;
– the presence of both mesangial and subendothelial deposits.
However, deposits are visible only if they are very abundant and, most often, mesangial proliferation and double-contour aspects dominate the picture. Proliferation often results in a considerable increase in the size of the glomeruli.
At the same time, the mesangial cytoplasm encircles totally or partially the glomerular capillary. This interposition considerably thickens the walls of the glomerular capillaries and contributes, with the mesangial proliferation, to sharply diminish the capillary lumen.
Other elements are also noted in light microscopy: the presence of polymorphonuclear cells in the capillary lumen, the presence of extramembrane deposits associated, sometimes voluminous in the form of humps, sometimes smaller, flattened on the basement membrane.
In electron microscopy:
Examination by electron microscopy allows a better definition of proliferation, thickening of the capillary wall and deposits.
– Aspect in double contour: it is the result of the mesangial interposition between two argyrophilic layers: the normal densa lamina on the external slope and strips of condensed mesangial substance on the internal slope. The endothelial cells are thus pushed back towards the lumen. The thickness of the interposed mesangial tissue can reach 60 times the thickness of the normal basement membrane. Various mesangial cell extensions, cell membrane remnants, matrix strands, and deposits are found.
– Deposits: subendothelial deposits, which may also be termed endomembranous, are in fact rather elongated along the basement membrane and separated from the endothelial cells by mesangial interposition. They may be small, segmental, sometimes voluminous, elongated and confluent, even thick and circumferential, as in lupic GNMPs, corresponding to the initial descriptions of the wire-loop aspects.
The mesangial deposits are sometimes of identical structure to the subendothelial deposits, sometimes denser, forming granular masses inside the mesangial matrix.
Subepithelial or extramembranous deposits are frequent: sometimes humps irregularly arranged along the capillaries, identical to those observed during acute poststreptococcal glomerulonephritis, sometimes smaller, separated from one another by extensions or spikes of the basal membrane. They resemble the characteristic deposits of extramembraneous glomerulonephritis.
– Epithelial cells: they are hypertrophied and have a frequent fusion of the pedicels. Their cytoplasm is vacuolized. In some cases they proliferate, forming a crescent. Elsewhere, there are synechiae between the glomerular basement membrane and Bowman’s capsule: they consist of more or less loose mesangial substance.
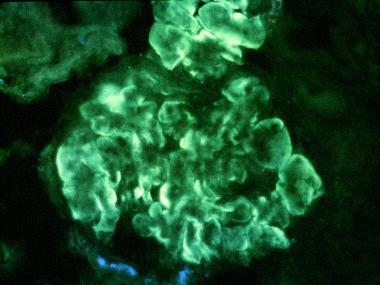
In immunofluorescence:
Immunofluorescence examination is used to determine the composition of observed deposits. In type I, it is very heterogeneous, as much as that of deposits observed in lupus glomerulonephritis. The deposits are at the same time peripheral, drawing the periphery of the lobules, and to a lesser extent, mesangial.
Two situations are schematically encountered in immunofluorescence, in idiopathic type I GNMP:
– In most cases, the deposits contain immunoglobulins and complement fractions Clq, C3, C4 on the periphery of the flocculus and also in the mesangium. There are cases in which, associated with the peripheral deposits of immunoglobulins and complement fractions, only deposits of C3 can be seen in the mesangium;
– Rarer cases have been grouped by the almost exclusive presence of both peripheral and mesangial C3 deposits while immunoglobulins and early complement factors are absent.
A variant is constituted by the presence of isolated mesangial C3, which some consider as a distinct entity. There is a controversy about this.
It is not unusual to find fibrinogen along the capillary walls, in the mesangium and naturally in the urinary chamber when there is extracapillary proliferation.
In secondary forms, the results of immunofluorescence may differ in the distribution and nature of the deposits: they are often atypical aspects in immunofluorescence which lead to the search for an associated disease (see etiological diagnosis below).
MEMBRANOPROLIFERATIVE GLOMERULONEPHRITE WITH DEPOSITS DENSES: TYPE II
GNMP with dense deposits within the basal membrane, or disease of dense deposits, is a precise anatomoclinic entity with only one morphological translation: the dense transformation of the renal basal membranes. It is also individualized by its close relations with the activation of complement by the alternate route.
In optical microscopy:
The main marker of the disease of the dense deposits is a banded, eosinophilic, refractive thickening of the basal membranes.
It is at the level of the glomerular capillaries that it is most marked, but it can also be demonstrated at the level of the Bowman capsule and in the tubular basal membranes. This thickening is very PAS-positive, due to an excess of sialic acid, dark green on the trichrome of Masson. It has a characteristic dark blue color on toluidine blue cuts, made after plastic inclusion. On colorations by silver, it typically stained in light brown. Although these membrane changes are very characteristic, they can be sporadic during the early phase of the disease.
In these cases, the diagnosis in light microscopy is uncertain and electron microscopy is necessary to clarify, unequivocally, the membrane modifications.
Apart from this dense transformation of the basal membrane, all the characteristic elements of type I GNMP can be observed in type II, which explains many confusions that lasted until around the 1970s. mesangial proliferation and increase of mesangial substance are less marked than in type I. There is no clear relationship between the extent of membrane damage and the degree of proliferation. As a corollary, the double-contour aspects are also inconstant.Proliferation may even be totally absent. In this case, some prefer to use the term “dense deposits disease” to that of GNMP.
In electron microscopy:
The dense, argyrophilous material, widening the lamina densa, called deposition, is in fact probably an alteration of the membrane structure: indeed, it does not present the granular composition usually seen in most deposits. It is often continuous, sometimes segmental, affecting more particularly the basal membrane adjacent to the mesangial axes.The same dense deposits are found in the mesangial matrix, the Bowman capsule, the basal membrane of the proximal circumferential tubes and, more rarely, the walls of the peritubular capillaries and the elastic plate of the arterioles. As in Type I, the subepithelial deposits observed, whether hump or smaller extramembrane deposits, have a finely granular structure and are not differentiated from those observed in acute poststreptococcal glomerulonephritis or extramembraneous glomerulonephritis, respectively.
Iterative biopsies usually show a decrease in mesangial proliferation as in Type I and, possibly, partial regression of dense deposits.
In immunofluorescence:
The appearance is homogeneous and most often allows a diagnosis of certainty. It is characterized by an almost exclusive fixation of the anti-C3 serum: the latter is weak, linear, continuous or discontinuous along the capillary walls, very bright on the contrary in the mesangium, forming nodules that are quite voluminous.
A closer examination, at a high magnification, shows that the membrane fixation of the anti-C3 is not homogeneous and rather appears as “rails” along the dense membrane material. A similar fixation is seen at the Bowman capsules and along the tubular basal membranes. Early components of the conventional Clq and C4 pathway are absent, as is properdin. Fibrin deposits are noted in the urinary chamber when there are croissants and may be present along the glomerular capillary walls.
TYPE III MEMBRANOPROLIFERATIVE GLOMERULONEPHRITE:
This third type has been individualized more recently in 1977, both by Anders et al and by Strife et al. It groups together aspects which, in light microscopy, could be classified as type I, but which show, in electron microscopy, zones of rupture of the basement membrane. These interruptions are due to a large accumulation within the basal membrane of granular deposits, often eosinophilic and refracting in light microscopy.
They are most often associated with endomembranous deposits and extramembrane deposits. Silver staining makes it possible to appreciate at best the extent of these basal membrane interruptions. Electron microscopy after silver impregnation can be used to distinguish Type III intramembranous deposits, negative from silver, from the argyrophilic thickening of the dense deposits: thus, as a whole, the basement membrane appears thickened, irregular, inhomogeneous, with deposits irregularly incorporated within it. The degree of mesangial proliferation is variable but generally much less than in type I. It is most often segmental.
In immunofluorescence, the deposits of C3 predominate as in the other types: they are noted throughout the glomerular surface, both in the capillary walls and in the mesangium.
Immunoglobulin G deposits are rarely encountered and always in small quantities. Clq and C4 are generally absent.
A controversy remains as to the individualization of type III as a distinct entity: thus, Habib and Levy consider it only as a variant of type I “insofar as, apart from its morphological characteristics, type III no clinical and immunological novelty “.
MISCELLANEOUS HISTOLOGICAL ASPECTS:
Lobular Glomerulonephritis:
For several years, it has appeared, thanks to the results of immunofluorescence and clinical studies, that it is only a morphological variety of GNMP. On the biopsies we can see all the degrees of lobularity. Lobulation is not related to either increased severity nor to an evolutionary stage of the disease: it is only a major hypertrophy of the mesangial substance, forming eosinophilic and PAS-positive nodules. These nodules are essentially composed of type IV collagen and laminin, while the bulk of the sclerotic mass between the collapsed loops contains type III and type IV collagen. At a very advanced stage, the appearance approaches nodular diabetic glomerulosclerosis.
A form of idiopathic nodular GNMP has recently been described, with voluminous PAS-positive deposits both mesangial and in the capillary lumens. It was not accompanied by cryoglobulinemia or lupus symptomatology. In immunofluorescence, there were essentially deposits of M and C4 immunoglobulins, associated to a lesser extent with C3 deposits and immunoglobulin A. Electron microscopy showed glomerular basal membranes of normal thickness, and a large quantity of finely granular subendothelial deposits, with no fibrillar or tubular structure.
Glomerulonephritis, membranoproliferative with extracapillary crescents:
In most cases of GNMP, the urinary chamber is free, although small synechiae can be noted between flocculus and Bowman’s capsule. In some cases, very rare, the presence of abundant epithelial croissants realizes the appearance of GNMP with croissants. These forms have been individualized within the heterogeneous group of diffuse crescent glomerulonephritis and have been distinguished, inter alia, by immunofluorescence, from poststreptococcal endo- and extracapillary proliferative glomerulonephritis. The initial clinical picture is, in both cases, rapidly progressive renal failure, but the subsequent natural course is much worse in the first case than in the second.
Glomerulonephritis, focal membranoproliferative:
Some glomeruli have the characteristic appearance of GNMP type I while others are normal, or have only moderate mesangial proliferation. It is in these focal forms that are encountered, among others, GNMP secondary to the ethyl cirrhoses.
In Type II, some focal forms are more commonly associated with acute nephritic syndromes than with nephrotic syndromes, with the most often normal complement and better progression of renal function than in diffuse forms.
Regardless of the type of GNMP, the presence of polymorphonuclear cells, the number of fibrous glomeruli, the number of crescents, and the severity of the associated tubulo-interstitial vascular lesions.
Differential diagnosis:
POSTSTREPTOCOCCIC ACUTE GLOMERULONEPHRITE:
It sometimes poses difficult diagnostic problems with GNMP. An acute nephritic syndrome, an increased antistreptolysin level, hypocomplementemia, mesangial proliferation with double-contour features and infiltration by polymorphonuclear cells may indeed be present in both cases. The absence of symptomatic regression, the persistence of the hypocomplementemia beyond the eighth week and, ultimately, the morphological evolution will decide, with appearance of characteristic features of GNMP on subsequent biopsies. However, the existence of certain relationships between GNMPs and acute poststreptococcal glomerulonephritis is retained.
OTHER DIFFERENTIAL DIAGNOSTICS:
Various problems may arise in the optical microscopy stage: they are generally removed by immunofluorescence examination.
Pre-eclampsia and thrombotic microangiopathy may have dual-contour features, as can other circumstances involving mesangial interposition.
Kimmelstiel-Wilson type diabetic glomerulosclerosis and light chain myeloma nephropathy sometimes have lobular aspects. The possible presence of intramembranous deposits can add an additional difficulty, giving an aspect close to type II. Some advanced extramembrane glomerulonephritis (III or IV) may be confusing with Type III GNMP.
Idiopathic lobular glomerulonephritis, or idiopathic nodular mesangial sclerosis, has recently been individualized by Alpers and Biava. It is seen outside of diabetes, amyloidosis, a disease of the light chains. The absence of significant deposits of immunoglobulins, complement in immunofluorescence, as well as the absence of electron dense deposits under electron microscopy distinguish it from GNMP. The symptomatology is rather insidious, associating a proteinuria often nephrotic and renal insufficiency. But as to its pathogenesis, we can not exclude the hypothesis according to which this lesion would represent the form of resolution of an anterior, subclinical glomerulonephritis.
Pathogenesis:
MEMBRANOPROLIFERATIVE GLOMERULONEPHRITE WITH SUB-ENDOTHELIAL DEPOSITS:
It is considered, although not shown, that Type I GNMPs are diseases associated with complement-activating immune complex deposition. A number of findings support this hypothesis:
– the presence of glomerular deposits of immunoglobulins and complement fractions;
– the detection, in glomerular deposits, of viral or parasitic antigens;
– frequent association with a hypocomplementemia indicating complement consumption. Indeed, the majority of patients have at least intermittent decreases in C3 concentrations and components of the classic complement activation pathway (C4, C1q);
– the presence of circulating immune complexes (CICs) or cryoglobulinemia in more than half of the cases;
– secondary causes of GNMP type I, particularly infectious, are frequently situations where there is a chronic antigenemia;
– the association of type I GNMPs with diseases mediated by CIC deposits, such as systemic lupus erythematosus. By analogy with the experimental model of serum sickness, it is possible that type I GNMPs are due to CIC deposits composed of antibodies and antigens derived from infectious agents on the internal surface of the wall of the glomerular capillary. The formation of CIC in situ is also possible, after initial fixation of the antigen on the glomerular basement membrane.
The presence of immune complexes in the mesangium and subendothelial spaces would trigger the activation of complement and the release of cytokines responsible for influx of cells from inflammation and proliferation of mesangial and endothelial cells.
The fact that most patients with CICs do not develop GNMP suggests the involvement of other factors in the pathogenesis of this type of glomerular nephropathy (size of immune complexes, nature of antigen, physicochemical characteristics of immunoglobulins, capacity purification of CIC by the reticuloendothelial system, local glomerular factors, etc.).
MEMBRANOPROLIFERATIVE GLOMERULOPATHY OF TYPE II:
According to West, excessive activity of C3 convertase alterne is responsible for the development of renal lesions observed in type II GNMPs. Indeed, in patients with type II GNMP, in more than 80% of the cases, there is a particular autoantibody, the nephritic factor C3 (C3Nef). By binding to the alternating C3 convertase (C3bBb), it protects this complex from enzymatic inactivation by factor H and leads to permanent activation of the alternate complement pathway. This translates biologically into a decrease in serum C3 concentrations. This hypothesis is supported by an animal model of pigs with factor H deficiency, hypocomplementemia and type II GNMP. In humans, the association of a factor H deficiency and a GNMP is also described. However, the mechanism responsible for glomerular lesions is not elucidated and serum concentrations of C3 convertase alternate were not measured in the West study.
Recently, Schwertz et al did not demonstrate elevation of C3bBb in 15 children with type II GNMP associated with the presence of C3Nef. Finally, a number of patients have progressive renal involvement, without modification of the complement fractions, independently of the presence of C3Nef.
If the intervention of C3Nef in the development of renal lesions is not obvious, its involvement in the lysis of adipose tissue observed during partial lipodystrophy, a condition commonly associated with type II GNMP, is much more direct.Indeed, complement seems to play a role in the regulation of adipose tissue. Adipocytes produce certain key fractions of the alternate complement pathway, such as factor D (also called adipsin), a protein that allows for the final synthesis of alternating C3 convertase. In 1993, Matthieson et al demonstrated that in the presence of C3Nef, membrane-mediated lysis of the adipocytes was observed. Another particularly informative element is the existence of regional variations in the expression of the factor D, which are superimposable to the lipodystrophy zones. One can imagine that the glomerular cells expressing complement fractions would be the target of the alternate C3 convertase.
However, to date, the chemical composition and nature of the dense deposits remain unknown. For some, type II GNMPs would be related to a primary disorder of the synthesis or degradation of the glomerular basement membrane, with complement anomalies being only a secondary phenomenon. Indeed, in cases of type II GNMP recurrence after transplantation, dense deposits appear before any C3 deposition, irrespective of the presence of hypocomplementemia and the presence of C3Nef.
Clinic:
The three morphological types of GNMP can not be distinguished clinically. Schematically, several tables can be individualized.
– In 50% of cases, it is an impure nephrotic syndrome of progressive installation.
– In 25% of cases, it is a typical acute nephritic syndrome following an otorhinolaryngological infection. It is sometimes superimposable in all respects, at least initially, on the table of acute poststreptococcal glomerulonephritis. The persistence of hypocomplementemia associated with proteinuria and hematuria beyond the eighth week is more evocative of GNMP.
– In 15% of cases, it is a discovery of systematic examination in an asymptomatic patient, showing proteinuria and microscopic haematuria. Intercurrent infections may be accompanied by episodes of macroscopic hematuria.
Another insidious presentation is that of arterial hypertension, at first moderate and then more difficult to control while proteinuria and microscopic haematuria remain very moderate.
Similarly, GNMP can be discovered in patients with chronic renal insufficiency of varying degrees, usually associated with proteinuria, microscopic hematuria and elevated blood pressure. Thus, GNMP can be latent for years and their revelation is only the result of a spectacular episode, nephrotic syndrome or acute nephritic syndrome.
– Immunologically, a characteristic trait of GNMP is the lowering of the serum concentration of total hemolytic complement and its component C3 (present in 75% of cases at a given time of the evolution). In fact, the hypocomplementemia is very often fluctuating and does not have a true correlation with the clinical picture. The complement activation is carried out by the classical route in type I and by the alternate route in type II. In the latter case, the intervention of a particular autoantibody, the nephritic factor C3, found in more than 80% of patients, is the cause of the continuous degradation of C3.
Prognosis:
The prognosis of GNMP is unfavorable since 50% of patients, children and adults, with nephrotic syndrome progress to end-stage renal insufficiency in 10 years. After 20 years, 80% of patients reach the stage of end-stage renal failure.However, in 10 to 15% of cases, complete and lasting remission is observed.
The presence of nephrotic proteinuria, arterial hypertension, severe interstitial lesions, extracapillary proliferation, and lobular forms are predictive factors for progression to end-stage renal disease. In their absence, the 10-year renal survival of Type I GNMPs is 85%.
Type II GNMPs are generally considered more aggressive than Type I GNMPs, progressing to end-stage renal disease within 5 to 12 years.
However, it is important to note that the etiological treatment of certain secondary forms, such as the eradication of a bacterial infectious focus, may be accompanied by clinical and histological remission.
Etiological diagnosis:
GNMPs may be idiopathic or secondary to a variety of diseases, including infectious or dysimmune diseases. Chronic HCV infections could account for up to 60% of cases of type I GNMP previously considered primitive.
DISEASES ASSOCIATED WITH MEMBRANOPROLIFERATIVE GLEMBERS OF TYPES I AND III:
Dysimmune Diseases:
Systemic lupus erythematosus:
Some forms of proliferative glomerulonephritis of lupus have a type I GNMP morphology with immunofluorescence deposits of immunoglobulins G, A, M and complement fractions (C1q, C4, C3), subendothelial and mesangial sites .The diagnosis is mainly provided by the clinical presentation and presence of native deoxyribonucleic acid (DNA) antibody in the serum.
Mixed Cryoglobulinemia Type II:
Most patients have HCV. However, some malignant haemopathies or autoimmune diseases (including Gougerot-Sjögren syndrome) may be accompanied by mixed cryoglobulinemia type II.
The observed GNMP has sub-endothelial and mesangial deposits of immunoglobulin G, but mainly of immunoglobulin M and complement. There is an important leukocyte infiltration of the flocculus, particularly rich in monocytes. The capillary lights are sometimes filled with occlusive hyaline thrombi. Vasculitis affecting the arteries of small and medium caliber is sometimes found. It is characterized by intraluminal deposits, fibrinoid necrosis of the arteriolar wall and perivascular leukocyte infiltration. The presence of extracapillary proliferation is rare. In electron microscopy, subendothelial or intracapillary deposits are either amorphous deposits resembling immune complexes, or deposits with a fibrillar structure organized into a tube (longitudinal section) and a cockade (cross-section).
Selective deficits of certain complement fractions:
A C2 homozygous deficiency appears to be associated with a higher frequency of GNMP compared to control populations. Sporadic cases of GNMP associated with a congenital C3 deficiency have been reported.
Infectious diseases:
They are the main causes of type I GNMP.
Bacterial infections:
Infection of a ventriculoatrial or ventriculoperitoneal derivative may result in GNMP (“shunt nephritis”). The germ in question is generally Staphylococcus epidermidis.
The table includes relapsed fever, haematuria, proteinuria, sometimes a nephrotic syndrome and renal insufficiency. A hypocomplemia is common. The removal of the infected shunt usually leads to complete, clinical and morphological healing. This situation, while becoming very rare, has the merit of demonstrating, in an exemplary manner, the reversibility of certain GNMP.
GNMPs have also been described in subacute bacterial endocarditis and in some chronic deep suppurations. In 1976, Beaufils et al reported a series of 11 patients with severe visceral infection and severe renal insufficiency. Only patients treated promptly and effectively recovered near normal kidney function.
Viral Infections:
· Hepatitis B
The presence of proteinuria or nephrotic syndrome during hepatitis B is accompanied by various glomerular lesions, ranging from extramembraneous glomerulonephritis to GNMP.
The mixing of the two sometimes achieves an aspect close to type III.
Cell proliferation is moderate. In immunofluorescence, the observed deposits contain immunoglobulin G, immunoglobulin M, C3 and sometimes immunoglobulin A. In some cases, mixed cryoglobulinemia is detected.
The HBs antigen has been directly recovered in many cases.
The efficacy of interferon in this situation has been reported.
· Hepatitis C
It is currently recognized that infection with HCV, and more particularly its association with mixed cryoglobulinemia type II, is one of the main causes of GNMP.
Analysis of HCV-associated GNMP observations showed that in nearly 90% of cases, mixed cryoglobulinemia was found (mixed cryoglobulinemia was found in 55-90% of HCV-infected patients, less than 1% kidney damage). These cryoglobulins can sometimes appear during evolution. In the Johnson et al study, if 14/34 patients had no mixed cryoglobulinemia at the initial stage, nine of them had secondary mixed cryoglobulinemia.
Initial work by Yamabe et al found no mixed cryoglobulinemia in six HCV-infected patients with GNMP, but in the subsequent publication, these six subjects had mixed cryoglobulinemia. The search for mixed cryoglobulinemia must therefore be repeated.
The possibility that GNMP is associated with HCV in the absence of cryoglobulinemia remains controversial. In France, the prevalence of anti-HCV antibodies was zero in 35 patients with a GNMP considered primitive. In an Italian study, only three of the 128 patients with idiopathic type I GNMP had HCV infection. Other studies in Spain, Turkey, Hong Kong, and the United States found no increase in the prevalence of HCV infection in subjects with primary GNMP.
Fornasieri et al demonstrated the particular affinity of monoclonal immunoglobulin M anti-immunoglobulin G produced by HC-stimulated lymphocytic B clones for fibronectin, a protein of the mesangial matrix. More recently, two teams have been able to localize HCV-specific antigens in the wall of glomerular capillaries and in the mesangium of patients with GNMP associated with mixed cryoglobulinemia type II. Finally, viral ribonucleic acid (RNA) is found in cryoprecipitate at concentrations 100 times higher, or even higher, than in serum. These arguments are in favor of a glomerulopathy mediated by immune complexes forming in situ or depositing itself via the immunoglobulin M j .
Parasitic infections:
· Bilharziasis
This is a non-exceptional cause of nephrotic syndrome in endemic areas. GNMP lesions, sometimes of type I, but more often of type III, have been described in the most severe forms of the disease. Glomerular involvement is observed in 10 to 15% of patients with hepatosplenic disease. Extracapillary croissants are sometimes associated.Intra- and extramembrane subendothelial deposits contain immunoglobulins, complement fractions and sometimes bilharzian antigens. In the hepatosplenic forms of the disease, the course of glomerular nephropathy is not modified by the antiparasitic treatment.
· Malaria
Kidney damage is not uncommon during quartan fever.
The glomeruli show varying degrees of mesangial proliferation, segmental sclerosis, thickening of the capillary walls, with double-contour appearance. Intra- and extramembrane subendothelial deposits contain immunoglobulins, complement fractions and sometimes plasmodial antigens.
Malignancies:
Malignant hemopathies are occasionally accompanied by nephrotic syndrome. It is usually an amyloid disease or minimal glomerular lesions, sometimes with focal glomerulosclerosis. In about 10% of cases, Type I GNMPs have been described, often in the presence of Type I or II cryoglobulinemia. Joint disappearance of the nephrotic syndrome and signs of chronic lymphocytic leukemia under immunosuppressive therapy may be observed.
In 1997, Ahmed et al published the case of a patient with nephrotic syndrome secondary to GNMP and renal adenocarcinoma. After surgical excision of the tumor, they observed a disappearance of proteinuria and an improvement of the renal function.
Hepatic impairment:
Nephropathy with mesangial deposits of immunoglobulin A is often encountered in the context of ethyl cirrhosis. It may have a focal GNMP appearance, with a particular feature: intense binding of the anti-immunoglobulin A serum, mesangial and subendothelial site.
In the literature, there are at least eight cases of a -1-antitrypsin deficiency, phenotype ZZ, with cirrhosis associated with a nephrotic syndrome. Renal involvement was most often GNMP, and in two cases glomerular deposits of α- 1-antitrypsin were found in immunofluorescence.
Sickle cell disease:
In homozygous forms, a number of glomerular disorders can be observed. True type I GNMPs with immunoglobulin G, M, C3 and C1q immunoglobulin deposits associated with the presence of immune complexes containing tubular epithelial antigen have been reported in the literature.
Splenorenal or hepatorenal “shunt”:
Dash et al. Have studied the influence of surgical derivation of portal circulation in 400 patients with portal hypertension secondary to non-cirrhotic hepatic fibrosis (FHNC), or an obstruction of the portal trunk of extrahepatic origin. At age 5, in patients with hepatic impairment with NHSC, 32% had nephrotic syndrome; more than half of these patients had GNMP (with a predominance of deposits of immunoglobulin A 2 nature). The group with normal liver function did not develop glomerular nephropathy, suggesting a lack of hepatic clearance of immune complexes aggravated by the shunt.
DISEASES ASSOCIATED WITH MEMBRANOPROLIFERATIVE GLOMERULONEPHRITES OF TYPE II: PARTIAL LIPODYSTROPHY
GNMPs with dense deposits can be observed in isolation or during a very particular condition, partial lipodystrophy.
This condition is characterized by the absence of adipose tissue in certain parts of the body, especially the upper limbs, trunk and face. Partial lipodystrophy is generally accompanied by hypocomplementemia and the presence of C3Nef. The association with retinal lesions (yellowish nodules in the fundus of the eye), secondary to dense deposits within the basement membrane of the pigment epithelium, is in favor of a systemic pathology.
Medical treatment:
The treatment of GNMP is far from being perfectly codified.
The analysis of published therapeutic results is difficult for a number of reasons: mostly uncontrolled retrospective studies, mainly including type I GNMPs, including evaluation criteria, treatment follow. On the other hand, these studies ignored the association between Type I GNMPs and HCV, which would account for more than half of the cases of Type I GNMP, previously considered primitive.
Among the different therapeutic approaches (anticoagulants, antiaggregants, non-steroidal anti-inflammatory drugs, corticosteroids, cyclophosphamide), only corticosteroid therapy in children and antiplatelet agents in adults have demonstrated efficacy in prospective, controlled and randomized studies.
The use of these treatments is mainly reserved for the idiopathic forms of GNMP, in the case of factors with poor prognosis (proteinuria greater than 3 g / d, presence of renal insufficiency, interstitial lesions …).
CORTICOIDS:
Children with idiopathic GNMP with proteinuria greater than 3 g / 24 h and renal insufficiency may in some cases respond to prolonged treatment with high-dose corticosteroids. In the Tarshish et al study of 80 children with GNMP (mainly type I), prednisone 40 mg / m2, 1 day out of 2 for 3 to 4 years, can slow down the progression to severe renal insufficiency. Thus, 10 years after initiation of treatment, 61% of patients treated with prednisone versus 12% of untreated patients had stable renal function.
Given the side effects of prolonged high-dose corticosteroids, fewer heavy protocols were tested. Ford et al conducted an uncontrolled study in 19 children with type I GNMP. After initial treatment with 2 mg / kg of prednisone 1 day out of 2 (3 to 4 months), they had a very high decrease up to a dose of 20 mg every other day, continued for several years.Initial hypertension in the majority of patients was medically controlled. After 3 to 10 years of follow-up, the majority of children had stable renal function and a marked decrease in glomerular lesions (systematic renal biopsy after 2 years of treatment).
The work of the Cincinatti group corroborates the interest of corticosteroid therapy at alternate days in the management of GNMP in children. This same group recently reported the limited interest of this therapeutic approach in type III GNMP.
In adults, there is no satisfactory study evaluating the efficacy of corticosteroids in the treatment of GNMP. However, in the light of pediatric studies, the use of prolonged corticosteroid therapy in patients at risk could be attempted.
IMMUNOSUPPRESSORS:
A study done more than 30 years ago by Kincaïd-Smith reported an improvement in renal survival in ten patients out of 16 with the combination cyclophosphamide, dipyridamole and anticoagulants.
In two subsequent controlled studies, one American, the other Australian, this benefit was not found. In the study of Cattran et al., Using the same treatment in 59 patients with GNMP, after 18 months of evolution, there was no significant benefit from the treatment.
PLASMAPHERESIS:
Their interest is little documented and very controversial. Their use in the case of florid extracapillary proliferation remains controversial.
ANTI-AGREGATE PLAQUETAIRES:
In a controlled study, Donadio et al evaluated the effect of aspirin (975 mg / d) and dipyridamole (225 mg / d) for 1 year in 40 patients with type I GNMP. (14% at 3 years) compared to the control group (47% at 5 years). However, this difference disappeared after 10 years of follow-up, suggesting the need to continue this treatment for several years. In a more recent study, Zauner et al tested the effect of aspirin-dipyridamole for 3 years in 18 patients with GNMP. Both groups had protein restriction and appropriate antihypertensive therapy. At the end of the study, there was no significant change in renal function from baseline; on the other hand, the group treated with platelet antiaggregants showed a clear decrease in proteinuria compared with the placebo group.
It is currently recognized that in adults with idiopathic GNMP, proteinuria greater than 3 g / d or impaired renal function, aspirin and / or dipyridamole therapy may be tried.
PARTICULAR CASES: MEMBRANOPROLIFERATIVE GLOMERULONEPHRITES ASSOCIATED WITH HEPATITIS C VIRUS
Only effective antiviral therapy can lead to decreased cryoglobulinemia and secondary renal damage. Over the past 10 years, numerous studies using interferon monotherapy have reported regression of renal manifestations under treatment. Unfortunately, there is usually a relapse when interferon is stopped. Rare observations on the effect of dual therapy with interferon-ribavirin in these patients have been reported. In view of the results obtained in patients with chronic hepatitis C treated with the interferon-ribavirin combination (nearly 50% prolonged remission), it seems justified to propose to these patients the antiviral treatment currently considered the most effective . In practice, the combination of interferon-ribavirin may be proposed for 18 months to 24 months (contraindication of ribavirin if creatinine clearance is less than 50 mL / min).
Finally, in the case of “florid” vasculitis in these patients with mixed cryoglobulinemia linked to HCV (acute renal failure, neurological manifestations), a “heavy” immunosuppressive treatment is first used, associating boluses of methylprednisolone, oral corticosteroids, cyclophosphamide and plasma exchange. Some uncontrolled studies found a clear improvement in renal function in these patients in 55-87% of cases. The antiviral treatment is introduced within 2 to 4 months after the initial treatment.
Transplantation:
MEMBRANOPROLIFERATIVE GLOMERULONEPHRITE WITH SUB-ENDOTHELIAL DEPOSITS:
Recurrence of type I GNMP after renal allograft occurs in 20-30% of cases. This recurrence leads to the loss of the transplant in 40% of cases, within an average of 40 months after the diagnosis. The risk of recurrence would be greater in case of transplant from a related living donor, and in a second transplant in case of recurrence on the first graft.
Ciclosporin would not have a preventive effect. The association of aspirinedipyridamole may stabilize renal function.
In optical microscopy, recurrence may be difficult to assert because of the lesional similarity with allograft glomerulopathy. However, immunofluorescence and electron microscopy make it possible to cut. In the case of recurrence, immunofluorescence constantly reveals large glomerular deposits of C3 (of immunoglobulin G and sometimes of immunoglobulin M), in contrast to allograft glomerulopathy where immunofluorescence mainly finds deposits of C- immunoglobulins M. In electron microscopy, there are endomembranous deposits in the case of recurrence, subendothelial clear spaces in the allograft glomerulopathy.
Type III GNMPs may also re-occur after transplantation.
In the only published case, the patient lost his transplant after 7 years.
MEMBRANOPROLIFERATIVE GLOMERULONEPHRITE WITH DEPOSITS DENSES:
In this case, the recurrence is almost constant. It is easier to affirm because of the “marker” constituted by the intramembranous dense deposits. The thickening of the basal membranes first appears at the level of the glomerular hilum in contact with the mesangium.
It is sometimes early, in the first weeks after transplantation, resulting in the appearance of non-nephrotic proteinuria during the first year. The recurrence leads to the loss of the transplant in 10 to 50% of the cases according to the series. There is no effective treatment. Plasma exchange could be interesting in severe forms.
As in Type I GNMPs, there is no relationship between persistence or occurrence of post-transplant hypocomplementemia and recurrence of the disease.
Despite the high frequency of recurrences of GNMP on the transplant, it remains justified to transplant these patients, since recurrences do not usually have a noticeable clinical impact.
Conclusion:
The concept of GNMP has evolved significantly since the initial descriptions and two major entities have emerged within many morphological and immunological subdivisions. On the one hand, type II, or disease of dense deposits, is characterized by a dense transformation of the basal membranes. On the other hand, GNMPs with subendothelial deposits, grouping together types I and III, appear as chronic diseases with immune complexes. In this group, secondary causes, particularly infectious, are the most frequent. Environmental changes may be at the root of the decreasing frequency of GNMP in Western countries. Finally, the use of effective antiviral treatments in patients with HCV-associated GNMP is a real therapeutic hope.