 Introduction:
Introduction:
Primary nephropathy with IgA deposits was originally described in 1968 by Jean Berger and Nicole Hinglais after the systematic study of renal biopsies in immunofluorescence.
Described first in France, it is currently the most common chronic primary glomerulonephritis in the world.Jean Berger had noted in his first description. For some of these conditions, including alcoholic cirrhosis and human immunodeficiency virus (HIV) infection, the presence of IgA nephropathy with histologic glomerular lesions is now clearly established, for other etiologies, hypothesis of a simple association has been mentioned.In lupus, IgA deposits frequently accompany IgG, C3, C1q, and C4, such as IgM.
The prognosis of primary nephropathy with IgA was long considered favorable, whereas at present IgA nephropathy is the leading cause of end-stage renal failure by chronic glomerulonephritis requiring the use of extrarenal purification techniques, and in France nearly 10 % of renal transplant patients have as initial nephropathy an IgA nephropathy.Progression to terminal renal insufficiency is now estimated at one-third of the cases.
Epidemiology:
Demographic and geographic data:
IgA nephropathy is observed at all ages, but clinical manifestations become most evident between 20 and 40 years of age. The male / female ratio ranges from less than 2/1 in Japan to more than 6/1 in Northern Europe and the United States. IgA nephropathy appears more common in Caucasian and Asian patients than in black patients. There are, however, disparities: its frequency is indeed very low among the Polynesians of New Zealand and very high among the Aborigines of Australia or the Indians of New Mexico.
Few studies have evaluated the incidence of IgA nephropathy.
In France, Holland, Germany, Italy, Denmark or the United States, the incidence varies from 10 to 40 new cases per million inhabitants per year. The prevalence of the disease is, in most studies, expressed as a percentage of cases of primary glomerulopathy or as a percentage of renal biopsies. Primary IgA nephropathy is the most frequent glomerulopathy in patients with renal biopsy in Asia (29.2%), Australia (12%), Europe (10.7%), and North America (3% , 5 to 5%). The disparities in global distribution are explained by a genetic influence but above all by a different policy of indication of the renal biopsy and of the detection of microscopic haematuria. In Japan, for example, urinary sediment abnormalities are systematically investigated in all schoolchildren and the presence of microscopic hematuria leads to a renal biopsy. In contrast, in the United States, Canada or England, renal biopsy is indicated only in the presence of abundant proteinuria and / or renal insufficiency. Japanese data suggest a high prevalence in the general population, as a systematic immunofluorescence study of live donor kidneys with no clinical manifestations resulted in mesangial IgA deposits in 16% of cases.
Immunogenetic Factors:
Family forms:
Although IgA nephropathy is most often a sporadic condition, familial forms have been reported.
The implication of a genetic factor in the pathophysiology of IgA nephropathy is also suggested by several observations: the ethnic and geographical disparities in the prevalence of the disease, the presence of familial clusters in and after renal transplants the deposits mesangial IgA recurrences in the transplant, making this glomerulonephritis a well-defined entity. Several synonyms are used for this glomerular lesion: Berger disease, IgA primary nephropathy, glomerulonephritis with IgA mesangial deposits, and abandoned today, glomerulonephritis with IgA-IgG mesangial deposits.
This glomerulonephritis with immune complexes, defined by the constant presence in immunofluorescence of mesangial deposits of IgA, is associated with varied glomerular histological lesions making classification difficult. There is, however, great clinical and biological heterogeneity of IgA nephropathies. Primary IgA nephropathies including Berger’s disease and, where there are extrarenal clinical signs, rheumatoid purpura and secondary IgA nephropathies should be distinguished. Rheumatoid purpura is developed in another chapter even if there are great similarities between these two diseases. For some of these conditions, including alcoholic cirrhosis and human immunodeficiency virus (HIV) infection, the presence of IgA nephropathy with histologic glomerular lesions is now clearly established, for other etiologies, hypothesis of a simple association has been mentioned. In lupus, IgA deposits frequently accompany IgG, C3, C1q, and C4, such as IgM.
The prognosis of primary nephropathy with IgA was long considered favorable, whereas at present IgA nephropathy is the leading cause of end-stage renal failure by chronic glomerulonephritis requiring the use of extrarenal purification techniques, and in France nearly 10 % of renal transplant patients have as initial nephropathy an IgA nephropathy.Progression to terminal renal insufficiency is now estimated at one-third of the cases.
Epidemiology:
Demographic and geographic data:
IgA nephropathy is observed at all ages, but clinical manifestations become most evident between 20 and 40 years of age. The male / female ratio ranges from less than 2/1 in Japan to more than 6/1 in Northern Europe and the United States. IgA nephropathy appears more common in Caucasian and Asian patients than in black patients. There are, however, disparities: its frequency is indeed very low among the Polynesians of New Zealand and very high among the Aborigines of Australia or the Indians of New Mexico.
Few studies have evaluated the incidence of IgA nephropathy.
In France, Holland, Germany, Italy, Denmark or the United States, the incidence varies from 10 to 40 new cases per million inhabitants per year. The prevalence of the disease is, in most studies, expressed as a percentage of cases of primary glomerulopathy or as a percentage of renal biopsies. Primary IgA nephropathy is the most frequent glomerulopathy in patients with renal biopsy in Asia (29.2%), Australia (12%), Europe (10.7%), and North America (3% , 5 to 5%). The disparities in global distribution are explained by a genetic influence but above all by a different policy of indication of the renal biopsy and of the detection of microscopic haematuria. In Japan, for example, urinary sediment abnormalities are systematically investigated in all schoolchildren and the presence of microscopic hematuria leads to a renal biopsy. In contrast, in the United States, Canada or England, renal biopsy is indicated only in the presence of abundant proteinuria and / or renal insufficiency. Japanese data suggest a high prevalence in the general population, as a systematic immunofluorescence study of live donor kidneys with no clinical manifestations resulted in mesangial IgA deposits in 16% of cases.
Immunogenetic Factors:
Family forms:
Although IgA nephropathy is most often a sporadic condition, familial forms have been reported.
The implication of a genetic factor in the pathophysiology of IgA nephropathy is suggested by several observations: the ethnic and geographical disparities in the prevalence of the disease, the presence of family clusters in certain populations, and the evidence of a significant binding between 6p22-23 locus and IgA nephropathy in 60% of familial forms whose genome was assayed for binding. Studies are underway to characterize the gene corresponding to this locus of susceptibility and to explain the phenotypic variations between individuals bearing the trait 6p22-23. Another trait of susceptibility has since been also identified, located on chromosome 3p23-24, supporting the hypothesis that the familial form of IgA nephropathy is a complex, multifactorial pathology where probably more than one susceptibility gene, associated with environmental factors, would be responsible for the development of the disease.
Other Genetic Studies:
Numerous studies have sought to show, in sporadic forms, genetic abnormalities explaining the individual predisposition to develop the disease. The first studies date back to 1976, a strong association is then described between the disease and the HLA antigen BW35.
Since then, other associations have been identified but the results of the different studies are sometimes discordant: the polymorphism of tumor necrosis factor alpha (TNF a ), interferon gamma (IFN- c ), interleukin 4 (IL 4), the renin-angiotensin system or CD14 is thus involved in the development or progression of IgA nephropathy.
Physiopathology:
The physiopathological mechanisms of Berger’s disease remain largely unknown. Until now there has been no animal model of the disease, limiting experimental studies, but the discovery of a small primate of the genus Marsh (Callithrix jacchus), spontaneously developing IgA nephropathy, should help to understand its pathophysiology . Recent research has focussed mainly on the study of immunoglobulin A1 (IgA1), which is the IgA subclass exclusively deposited in the mesangium.
This mesangial IgA1 contains piece J in the absence of the secretory component. Mesangial IgA1 is polyclonal but more frequently contains the lambda light chain than kappa.
Reminder on the IgA system:
IgA immunoglobulins are the main antibodies produced by the mucosal immune system. This is called MALT for mucosal associated lymphoid tissue, grouping the immune system of the digestive mucosa (GALT, gut associated lymphoid tissue), bronchopulmonary (BALT, bronchial associated lymphoid tissue), nasal (NALT, nasal associated lymphoid tissue) Waldeyer ring. The IgA system in humans consists of two subclasses, IgA1 and IgA2, which differ by 22 amino acids from the alpha heavy chain. The hinge region, deleted in part on IgA2, is rich in serine bearing O-glycosylated residues. Their distribution, serum and in the various secretions, varies according to the isotype. Thus, 90-95% of serum IgAs are IgA1, 60% of IgAs in the colon are IgA2 and 90% of IgAs in the nasal mucosa are IgA1.
The structure and properties of IgA make it a particularly suitable system for protecting the body from infections.
Indeed, the association of one to two dimers of IgA to the molecule of the secretory component allows its transport in the intestinal lumen. Its oligomeric nature facilitates its high affinity interaction for the viruses and bacteria present in the secretions. IgA can not bind complement by the classical route but they represent the most effective immunoglobulin isotypes to activate the alternate route. IgA are not sensitive to the action of proteolytic enzymes, especially in the digestive tract where proteases are present at very high concentrations. Finally, the particular affinity of these immunoglobulins for mucus would reinforce its role as an obstacle to the penetration of pathogens through the digestive mucosa and the presence at the end of the IgA alpha chain of oligosaccharides containing mannose inhibits the adherence of bacteria to epithelial cells.
IgA is locally synthesized in the mucosal epithelia, where their level is elevated as opposed to their serum level, low compared to that of IgG. Adult levels of serum or serum IgA are reached as early as 7 and 10 years of age. They can be increased during liver disease, especially those complicated with cirrhosis.
Pathophysiology of Berger’s disease:
Role of the IgA system:
The existence of a circulating serum factor at the origin of Berger’s disease is supported by clinical observations. IgA deposits rapidly recur on the mesangium of a transplanted kidney in more than 30% of end-stage IgA renal transplants for IgA. In contrast, after kidney transplantation of a kidney with Berger’s disease in a recipient with another renal disease, the deposits disappear. Analysis of serum and renal biopsies in patients with Berger’s disease, such as rheumatoid purpura, suggested the pathogenic role of IgA itself, and in particular immune complexes containing IgA.
Immune complexes containing IgA:
In one half of the patients, there was an increase in the serum IgA level, which was unbalanced in favor of the IgA1 subclasses. These immunoglobulins are polymeric, unlike the general population, and the study of their molecular weight, often found at more than 103 kDa, suggests that they are, at least in part, circulating immune complexes composed of IgA. These immune complexes composed of IgA activate the alternative pathway of complement, as demonstrated by colocalization, in immunohistochemistry of IgA and the C5b-9 complex and the absence of C1q in the injured tissues, whereas the serum levels of the fragments C1, C3, C4 of the complement are normal. No specific antigen of the disease and common to all patients could be identified. The hypothesis of an overall increase in the synthesis of IgA, at the origin of the formation of the immune complexes was thus favored.
IgA production abnormalities:
The production site in excess of the IgA has been debated for a long time. There is certainly an excess of production of dimeric IgA in the mucosa of the tonsils but it does not appear that a simple excessive production of IgA by these mucous membranes can be at the origin of the mesangial deposits since, on the contrary, one observes to this level decreases the secretory IgA response. The predominance of IgA1 and the relative absence of IgA2 in the mesangium would be more in favor of medullary overproduction. In the peripheral blood of patients with Berger’s disease, more IgA-specific T-helper cells were observed in comparison with subjects not carrying this nephropathy and these cells spontaneously produced more interleukin-2 in vitro.
But, the increase in the amount of IgA alone in these patients can not explain the symptomatology. Indeed, other pathologies, such as IgA myeloma or HIV infection with hypergammaglobulinemia, are accompanied by excessive production of IgA and are exceptionally complicated by IgA nephropathy.
Anomalies of IgA clearance regulation:
There are abnormalities in the glycosylation of IgA, in particular the galactose and sialic acid content of IgA1, which would be responsible for a decrease in their clearance but also facilitate their deposition in the capillaries by increasing their affinity for different components of the IgA1. mesangial matrix. A deficiency of b 1-3 galactosyltransferase would be at the origin of this hypogalactosylation. Finally, in the absence of galactose, the terminal sugar, N-acetylgalactosamine, or the glycopeptide of the hinge region, could be recognized by the glycans-specific anti-IgA1 or IgG antibodies, thus forming the complexes
immune circulatory diseases.
Abnormality of IgA binding to its receptor:
The classically recognized IgA receptor is CD89 or RFc a I but is not expressed by the mesangial cells.
The mesangial receptor of IgA has recently been identified as the transferrin receptor or CD71. The expression of the transferrin receptor (RTf) binding IgA1 to the surface of the mesangial cells seems to correlate with the intensity of the cellular proliferation. However, its pathogenic role during the disease is not yet clearly demonstrated. One hypothesis would be that the binding of hypogalactosylated IgA to RFc would result in cleavage and release of the extracellular portion of the receptor. This would have two consequences:
• decreased membrane expression of RFc a I on circulating monocytes, responsible for decreased clearance of polymeric IgA;
• the released soluble RFc binds the serum polymeric IgAs forming circulating IgA-RFc complexes to I. These complexes would deposit in the kidney by binding to RTf, the expression of which on the surface of the mesangial cells is increased.
Hyperactivity of the mucosal immune system:
It has been suggested that a particular sensitivity of the mucosal immune system exists, dependent on IgA since the digestive permeability to antigens is increased, as is the number of B lymphocytes in the mucosa and tonsils.
Moreover, macroscopic haematuria is often preceded by an infection of the ORL sphere.
Consequence of IgA binding to the mesangial receptor:
Activation of mesangial cells by immune complexes containing IgA is considered the initiating event of IgA nephropathy. In vitro, these immune complexes stimulate the production of proinflammatory mediators such as cytokines (IL6, IL1), chemokines (IL8, macrophage-inflammatory protein [MIP], interferon-inducible protein [IP-10] (TNF- a , transforming growth factor b [TGF- b ]) capable of inducing mesangial cell proliferation or increasing the extracellular matrix. In vivo, the amount of IL6 in urine, the tubular and interstitial expression of type 1 and intrarenal adhesion molecules of proinflammatory cytokines and chemokines is correlated with the severity of renal involvement and would be a prognostic factor. The progression of renal lesions to glomerular sclerosis, tubular atrophy and interstitial fibrosis is then variable from one patient to another, dependent as for any other nephropathy, on its genetic background.
Clinical Manifestations:
In the early stages of the disease, 30-40% of patients have no symptoms and the diagnosis is suspected on systematic urinary examinations (eg occupational medicine) or another pathology.
Generally, clinical signs, when present, are not very specific. Classically, patients with Berger disease present episodes of macroscopic hematuria, contemporaneous with upper airway infection (within 24 to 72 hours). In patients with no clinical symptoms, proteinuria and microscopic hematuria may persist at a moderate and fluctuating flow.Nephrotic syndrome is rare but can be observed at all stages of the disease, early as terminal, then it probably has a very different meaning. Acute renal insufficiency is exceptional, occurring during crescent IgA nephropathy or contemporary episode of acute tubular necrosis macroscopic hematuria. Finally, hypertension is common in these patients, even in the absence of renal insufficiency. Sometimes the patient presents with a table of malignant hypertension which often reveals advanced nephropathy, evolving silently for a long time. It is exceptionally associated with biological signs of hemolytic uremic syndrome (HUS), requiring a genetic study in this case.
The indication of the renal biopsy, the only way to make the diagnosis, is still the subject of debates. Some advocate it only if the proteinuria is greater than 1 g / 24 h or if there is a renal insufficiency. The major defense argument of this attitude is the lack of treatment of the disease at an earlier stage.
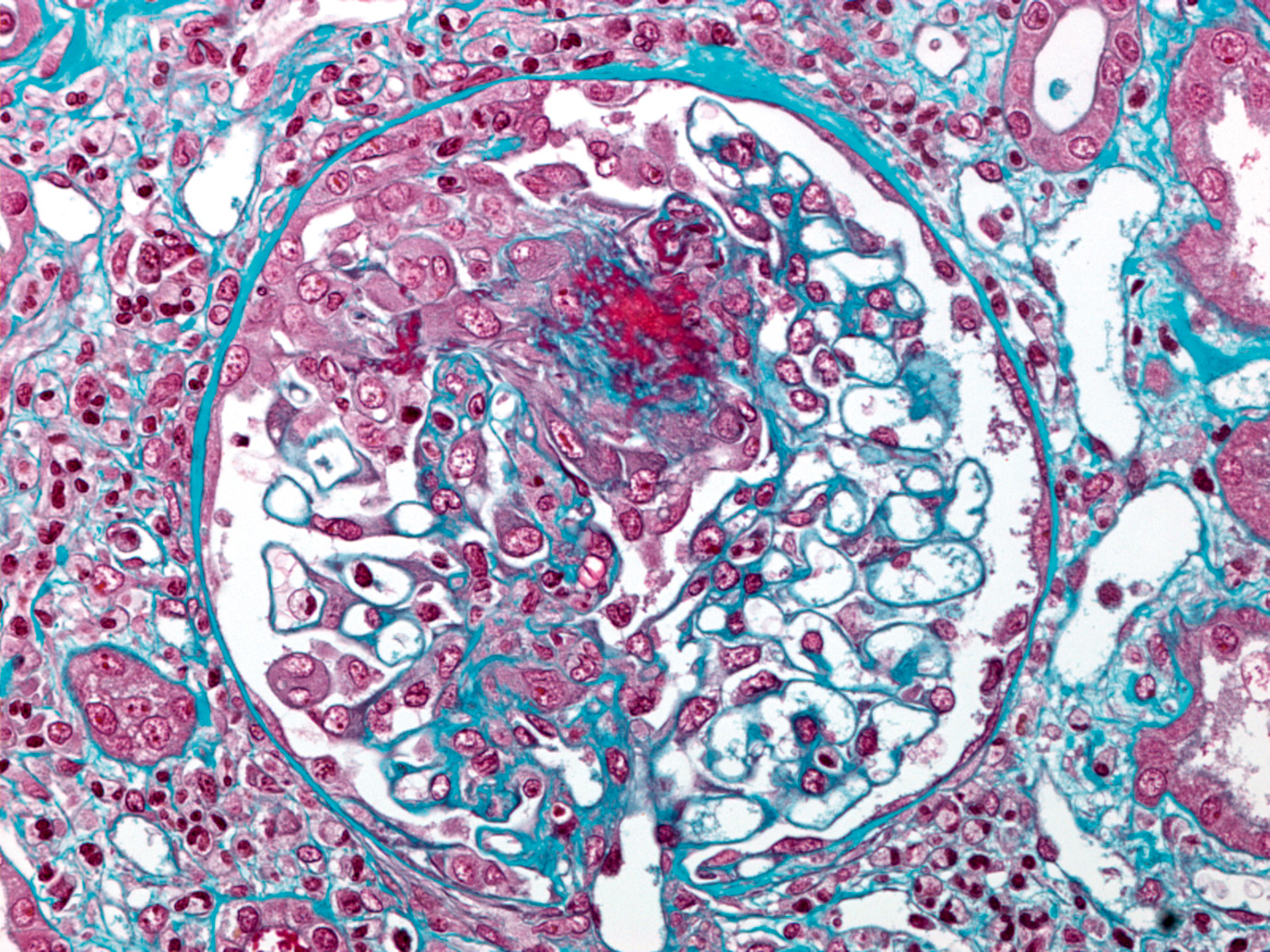
Histopathology:
Immunofluorescence:
The diagnosis of IgA nephropathy requires an examination in immunofluorescence (IF) with evidence of constant and predominant mesangial IgA deposits in the form of granular or filamentous marking of the mesangial areas in ” branch of tree “or” fingers of the hand “. They are observed in all glomeruli, isolated or associated with other immunoglobulins and complement factors. Present in the lactic region of the vascular pole, they are absent in the wall of pre-glomerular arterioles. In the highly proliferative forms of mesangial glomerulonephritis, they overflow into the glomerular walls, becoming mesangioparietal. One of their peculiarities is to be identifiable in the sclerotic glomeruli of the terminal kidneys allowing a retrospective diagnosis of glomerulonephritis even at this stage of evolution.
IgA deposits are associated in 40% of cases with IgG deposits and in less than 20% of cases of IgM. The C3 fraction of the complement is constantly present in the mesangial deposits and often has a more finely granular appearance than the IgA, it is also found in the arteriolar walls. The C5b-C9 membrane etching complex, properdin, factor B and factor H may also be present. The presence of fibrin / fibrinogen is found in 10 to 15% of the cases, in the same mesangial localization. Glomerular segmental lesions when necrotic fix fibrin. Segmental lesions of segmental and focal hyalinosis (HSF) type, often fibrohyalins fix IgM, C3 and C1q.
Optical and electronic microscopy:
Mesangial lesions:
Glomerular lesions are extremely variable within the biopsy. The glomeruli may appear normal or be the site of an enlargement of the mesangial axes by increase of proteins of the extracellular matrix (fibronectin and collagen IV). It is possible to identify “red” rounded deposits of fibrinoid aspect in the trichrome, in the mesangial position and in the wall, sometimes lifting the peripheral capillary wall. The non-proliferative forms correspond to the mesangiopathic forms.Mesangial hypercellularity is frequent, of unequal intensity from one glomerular lobule to the other. An intense mesangial proliferation can result in the formation of double segmental contours.
Much more rarely, and more commonly in children, there is diffuse endocapillary glomerulonephritis affecting all glomeruli or even more rare an endocapillary and extracapillary glomerulonephritis with epithelial crescents that may be circumferential in more than 50% of the glomeruli.
In the acute phases, inflammatory cells, including polynuclear cells, exist in the glomeruli. Similarly, focal thrombosis and foci of segmental fibrinoid necrosis of the flocculus can be observed.
Diffuse Mesangiopathic and Diffuse Proliferative Mesangial lesions are present only in 20% of cases. It is the segmental and focal forms that represent the majority of the histological forms of Berger’s disease.
Other superfluous glomerular lesions:
During the evolution of this chronic glomerulonephritis subjected to hematuric outbreaks of multiple segmental lesions appear. Some are present at the outset, very active in the form of focal thrombosis or foci of fibrinoid necrosis complicated by an endo and extracapillary segmental proliferation in crescent. These lesions evolve towards the destruction of a part of the flocculus with fibrohyaline scar producing synechia (often multiple synechiae within a glomerulus) between the flocculus and the Bowman capsule. Necrotic lesions may be “explosive”, revealed by macroscopic haematuria and acute renal failure, with biopsies of fibrinoid necrosis or blood in the urinary chamber of glomeruli surrounded by hematic cylinders. Finally, in proteinuric forms, there are typical glomerular lesions of segmental and focal hyalinosis and podocytic lesions.
Tubulo-interstitial and vascular lesions:
Tubulo-interstitial lesions are frequent in segmental forms, with spans of fibrosis, containing atrophic tubes and hematic cylinders during outbreaks.
Inflammatory cellular infiltration by lymphocytes, macrophages, and more rarely lipophages can be important.
It is a glomerulonephritis which is complicated by an arteriosclerosis of the interlobular arteries observed in the majority of the biopsies of young adults. Finally, there are specific vascular lesions in patients developing end-stage renal disease with severe or malignant arterial hypertension. These are lesions of thrombotic microangiopathy affecting pre-glomerular arterioles and small interstitial arteries, the other arteries showing severe arteriosclerosis.
Histological classifications:
Due to the great diversity of histological lesions during IgA nephropathy, numerous classifications have been proposed and evaluated for their ability to predict the course of renal disease, most often in terms of renal survival.
Semi-quantitative scoring systems, such as those used by Alamartine or Radford teams, assign a semi-quantitative grade (eg 0 to 4 for severe absence) to each glomerular, tubular, interstitial and vascular lesion that , with a histological score ranging from 8 to 20. The advantage of this type of classification is to identify morphological changes most correlated with clinical prognosis, but they are time-consuming and rarely used in common practice. Simpler classifications, limited to glomerular, tubulo-interstitial or chronic histological lesions, have also been developed.
The most commonly used are those of Lee, Haas and the World Health Organization classification for lupus nephropathy.
In our group, we use in our group the classification proposed by Niaudet for rheumatoid purpura and Berger’s disease in children to type glomerulonephritis in segmental or proliferative form, to which is added the details of the active lesions (hypercellularity, fibrotic necrosis, HSF, cellular growths, hematic cylinders, vascular lesions of thrombotic microangiopathy [MAT]), some of these lesions being susceptible to treatment, and chronic lesions (PAC, interstitial fibrosis, atrophic tubes, synechiae and fibrous growths , arteriosclerosis, cicatricial MAT), having more prognostic value.
Evolution and prognostic factors:
Complete remission, without proteinuria or urinary sediment abnormality, is observed in less than 10% of cases.Conventionally, IgA deposits in immunofluorescence never disappear. Renal insufficiency progresses slowly to become terminal in 25 to 50% of patients at 20 or 25 years depending on the series.
Numerous prognostic factors, both clinical and histological, were evaluated with contradictory results according to the authors and the methodology used in these studies. Indeed, the identification of predictive factors for the progression of renal insufficiency is necessary to isolate patients who could benefit from more aggressive treatments, especially within the framework of therapeutic protocols. Many studies are difficult to interpret because they contain a population that is too small in number, not always homogeneous, and too brief to follow. Nevertheless, clinically, age, persistence of a high proteinuria flow, renal function and blood pressure level are the recognized risk factors for progression of renal insufficiency.
Histologically, glomerular sclerosis and interstitial fibrosis are two predictors of poor prognosis. The presence of a class I or V according to the classifications of Lee or Haas, is more disputed.
Evolution of the disease varies geographically, probably due to different management over time: the delay between diagnosis and end-stage renal disease is shorter in patients with late onset of chronic renal failure , than in those who were diagnosed with isolated microscopic hematuria detected in systematic examinations. Nevertheless, caution should be exercised in relation to these prognostic factors, which do not appear to be applicable at the individual level.A Japanese study, confirmed by a Chinese study, reported 72 patients presenting with a diagnosis of normal renal function and a low rate of proteinuria, less than 0.4 g / 24 h. After 7 years of follow-up, proteinuria was greater than 1 g / 24 h in one third of patients, 26% became hypertensive and 7% degraded their renal function. In contrast, some patients with persistent nephrotic syndrome retain normal renal function after many years without treatment.
Recently, new progression markers have been studied, including genes involved in the activation of mesangial cells and monolymphocytic cells (CD34, CXCR3) in the renin-angiotensin system, adhesion molecules (ICAM-1, ELAM -1, VCAM-1), cytokines or chemokines (PDGF, IL1, IL6, IL10, FGF, HGF, TGF b ), and finally mediators of inflammation, sclerosis and apoptosis.
No molecule in this exhaustive list appears to be a specific marker of progression of IgA nephropathy.
Recurrence on the renal graft:
Renal transplantation provides an excellent therapeutic option for patients with end-stage IgA nephropathy. But mesangial deposits of IgA recur in glomeruli of the graft after renal transplantation in nearly 50% of cases. It is demonstrated between 0.3 and 213 months (on average 40 months) after transplantation, of course depending on the indication of the biopsy (no systematic biopsy). Graft dysfunction due to recurrence of initial disease is estimated to be 13% at 5 years and graft loss between 2 and 16%. Although this rate of graft loss is low, with IgA nephropathy being the most frequent glomerulopathy of end-stage renal disease in the world, the number of patients requiring a second transplant is increasing, especially as these patients are youth. Recurrence appears to be more important when the graft is from a living living donor, although this notion is disputed. The use of ciclosporin or mycophenolate mofetil does not seem to influence the frequency of recurrences.
Pregnancy:
Several studies have demonstrated that IgA nephropathy in a woman with preserved renal function, moderate proteinuria and low or no arterial hypertension has no adverse effect on the proper course of pregnancy for both the mother and the child. for the child. In contrast, if the mother does not meet all of these conditions, pregnancy should be closely monitored because the risk of obstetric complications is high.
Treatment:
Until recently, no effective treatment was available to treat IgA nephropathy. Even in the absence of curative treatment, a number of therapeutic options proposed in recent years appear to be effective in slowing down the progression of nephropathy. Nevertheless, the extreme variability in the clinical course of IgA nephropathy and its very slow rate of progression make it very difficult to evaluate therapeutic clinical trials, which in fact require, and rarely do, a large number of patients prospectively for a very long time. It should also be emphasized that very few therapeutic studies take into account the histological criteria to include patients. A classification recognized by the community as a whole and used in all future studies would, if possible, assess the effectiveness of treatments within a more homogeneous population. The therapeutic attitude most often recommended for the management of a patient suffering from IgA nephropathy.
Hygiene measures:
As with any renal disease, the patient should be given a tobacco withdrawal and the use of non-steroidal anti-inflammatory drugs should be avoided. The blood pressure objective, if the proteinuria is greater than 1 g / d is 125/75 mmHg.
Blockage of the renin-angiotensin system:
Among the antihypertensive treatments, blockers of the renin-angiotensin system are recommended as first-line therapy. Several studies have demonstrated their effectiveness, more than any other class of antihypertensive drug, in slowing the progression of renal insufficiency during IgA nephropathy:
• enalapril, which preserves renal function and decreases the proteinuria output independently of the blood pressure in a prospective, placebo-controlled study over 6 years, including 44 patients with Berger disease with preserved renal function but one proteinuria rate greater than 0.5 g / d;
• In contrast to amlodipine, other converting enzyme inhibitors (temocapril and trandolapril) have been shown to reduce renal dysfunction in 49 patients with Berger’s disease, regardless of the amount of tubulo-interstitial fibrosis present on renal biopsy at diagnosis;
• losartan, more than amlodipine, decreases urinary excretion of TGF- b 1 and proteinuria at equivalent blood pressure level after 12 weeks of treatment in 38 patients with IgA nephropathy, the proteinuria ratio is between 1 and 3 g / d;
• valsartan administered for 104 weeks to 54 patients with IgA nephropathy and hypertensive patients with a proteinuria rate greater than 1 g / d and moderately altered renal function defined by creatinine between 120 and 250 μmol / l significantly decreased decreased proteinuria and slowed renal degradation;
• finally, during the COOPERATE study, it was shown that the association of an angiotensin 2 receptor antagonist with an inhibitor of the conversion enzyme increased the renoprotective effect by further reducing the flow rate of proteinuria, always independently of the tension control in many chronic nephropathies, including 131 diseases of Berger.
Corticosteroids:
A meta-analysis of six available studies of sufficient quality suggests the efficacy of corticosteroid therapy to reduce proteinuria and the risk of progression to end-stage renal disease. Only two studies are prospective and include a significant number of patients.
The Italian study, with 10 years of follow-up, shows impressive results in terms of renal survival (97 versus 53%) in the group of patients with Berger’s disease with normal renal function and proteinuria rate between 1 and 3, 5 g / d, treated with 1 g / d methylprednisolone for 3 consecutive days, repeated in the second and fourth months, and 0.5 mg / kg / day between treatments. It should be noted, however, that few patients, although equally distributed in each group, received treatment blocking the renin angiotensin system and especially only 12 of the patients treated and 7 of the untreated patients of the 43 patients included at the start of the study in each group are still present at the end of the study.
Another recent study included 43 patients with Berger’s disease with normal or moderately altered renal function and moderate histologic lesions according to their own criteria for histological activity and treated with a low dose of steroids (20 mg / day progressively decreased over 2 years) and dipyridamole. The 47 patients in the control group received only dipyridamole. It also demonstrates the ability of steroids to reduce the flow of proteinuria but the lack of effect in terms of renal survival.
Examination of renal biopsies of 16 patients before and after 12 months of moderate-dose corticosteroid therapy showed a reduction in mesangial matrix accumulation.
Finally, a single study, carried out in 34 patients, evaluated the effect of steroids in the presence of a free nephrotic syndrome and demonstrates their efficacy only in the histological presence of normal or subnormal glomeruli.
Immunosuppressants:
Cyclophosphamide associated with corticosteroids:
A single study shows the superiority of an immunosuppressive treatment including corticosteroid-associated cyclophosphamide to decrease proteinuria and improve renal survival. This study included 38 patients with IgA nephropathy with renal dysfunction defined by creatinine between 130 and 250 μmol / l after blood pressure control (<160/90 mmHg). Patients were treated with placebo or corticosteroids combined with oral cyclophosphamide at a dose of 1.5 mg / kg / day for 3 months, followed by azathioprine at the same dose for 2 years. Renal survival of treated patients versus placebo-only patients was 82 versus 68% at 2 years and 72 versus 26% at 4 years. The study focuses on a small
series of patients, and especially the definition of blood pressure has since been revised downward. Finally, the use of treatment blocking the renin-angiotensin system is unspecified.
Mycophenolate mofetil:
Three studies evaluated the benefit of mycophenolate mofetil treatment during IgA nephropathy. The first showed no superiority of this treatment compared to a placebo, in a small number of patients at risk of progression, combined with a treatment with an enzyme-converting inhibitor and a salt-free diet. The second study, conducted in 20 patients with more than 1 g / d of proteinuria despite the use of a renin-angiotensin blocker and normal or moderately altered renal function, highlights the ability of mycophenolate mofetil to reduce proteinuria more than placebo. The last one included 32 patients with IgA nephropathy with more than 1 g of proteinuria and either arterial hypertension or renal dysfunction defined by a creatinine clearance of between 20 and 80 ml / min and finally the presence on renal biopsy of glomerulosclerosis or interstitial fibrosis. No benefit of mycophenolate mofetil is observed in this population. The small number of patients in these studies does not allow us to conclude definitively on the efficacy of mycophenolate mofetil, two prospective studies are in progress.
Fish oils:
Concentrated fish oils contain a high proportion of omega 3 fatty acids, including eicosapentaenoic acid, and would have the ability to decrease the production or action of cytokines induced by the inflammation involved in the progression of renal lesions.
A single study in 106 patients with IgA nephropathy with abundant proteinuria showed superiority of this treatment over a 2-year period compared with placebo to slow down even when the treatment was discontinued. renal function, irrespective of the rate of proteinuria, and especially as renal function was impaired. Patients included in this study had advanced nephropathy with degradation of renal function (creatinine> 132 μmol / l) and high proteinuria (2.5 to 3.2 g / d). The main criticism of this study is that in the placebo group (olive oil) the rate of progression was faster than is generally observed in other studies.
A more recent study does not seem to confirm these results: the patients included all had a glomerular filtration rate (GFR) greater than 50 ml / min / 1.73 m 3 and a moderate to severe proteinuria. Thirty-two patients received corticosteroids alone, or omega-3 fatty acids or placebo.
After 2 years, 72% of the patients are still present in the study.
There is no benefit in terms of renal survival of steroids or omega 3 fatty acids. Finally, it should be noted that this treatment is costly (not repaid in France) and compulsory (numerous tablets) and has as a side effect a malodorous breath often poorly tolerated by the patient.
Tonsillectomy:
Exacerbation of urinary symptoms in upper respiratory tract infections, especially in the tonsils, has led some authors to recommend tonsillectomy in patients with repeated macroscopic hematuria episodes, especially as several studies evidence of local anomalies in the production and metabolism of IgA (see above chapter Physiopathology).
Two Japanese retrospective studies, including a large number of patients, show a rapid benefit in terms of decreased proteinuria output and after more than 10 years in terms of renal function. A prospective study to compare the use of corticosteroids with or without tonsillectomy is underway in Japan.
Recommend to treat any infectious outbreak to avoid possible outbreaks of the disease seems reasonable even in the absence of published study supporting this attitude.
Special case of IgA nephropathy with extracapillary proliferation:
IgA nephropathy with a rapidly progressive clinical manifestation of glomerulonephritis in relation to the presence of extracapillary proliferation is very rare but very poor prognosis even more than other glomerulonephritis with increasing growth. Several isolated cases and a series of 12 patients with rapidly progressive renal failure and extracapillary proliferation to renal biopsy have been reported.
A treatment combining cyclophosphamide boluses and corticosteroids for 6 months resulted in a 36-month decline in the proportion of end-stage renal disease in the treated group. However, a large randomized prospective study is needed to determine the effectiveness of this type of aggressive treatment.