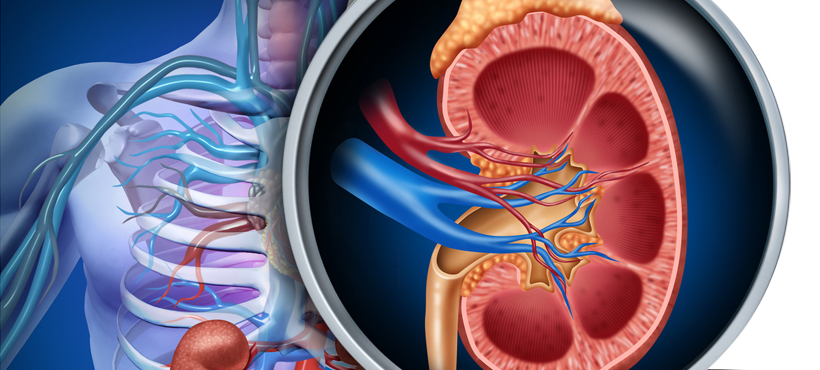
AUTOSOMIC RENAL POLYKYSTOSIS DOMINANT:
 Characterized by renal cysts possibly associated with liver cysts and cardiovascular abnormalities, autosomal dominant polycystic kidney disease (ADPD) is an inherited disorder that usually occurs in adulthood.
Characterized by renal cysts possibly associated with liver cysts and cardiovascular abnormalities, autosomal dominant polycystic kidney disease (ADPD) is an inherited disorder that usually occurs in adulthood.
Epidemiology:
PRAD is the leading cause of kidney disease, with prevalence estimated at 1 per 1,000 in Caucasian populations. In Western countries, it causes 5 to 10% of dialysis patterns. This incidence is 3% in Japan and 21% in Iceland.
Two genes are responsible for the vast majority of PRADs: PKD1, located on the short arm of chromosome 16, is involved in about 85% of cases; PKD2, located on chromosome 4, is responsible for most other cases. Neomutations have appeared rare (about six cases out of 300) in Wales, where this has been well studied.
Kidney manifestations:
PRAD can be discovered during kidney or extra-renal complications, a family survey, or accidentally on the occasion of an ultrasound or even a CT scan. The renal semiology includes the facts specific to the cysts (pain, hematuria, infection, calculus) and the impact on the functioning of the kidney (hypertension and renal insufficiency).
Pain:
They reveal the disease about once in four; their frequency increases with the age of the patients and the size of the kidneys. Chronic heaviness is common, especially when the volume of the cysts is large. Daily and prolonged use of analgesics is not desirable since these products are probably nephrotoxic. Although a publication once again emphasized the improvement from surgical decompression of the largest cysts without any evidence of renal impairment, this perilous surgery should only be considered exceptionally. Acute pain is caused by intracystic bleeding; more rarely by urinary obstruction by calculation or clot: the ultrasound is indicated in case of intense or prolonged pain to objectify a possible dilation of the excretory way, sometimes difficult to assert because of the reworkings related to the cysts. If pain and fever coexist, an infection is likely.
Hematuria:
Spontaneous or post-traumatic, it is attributed in the absence of computation at the rupture in the excretory way of vessels sent back by the walls of the cysts. Endo-urological explorations should be avoided. Abundant drinks are helpful in reducing the risk of clot obstruction. The bleeding rarely lasts. Its persistence requires bed rest. Microscopic hematuria is observed in 25% of patients with PRAD.
Exceptionally long-lasting bleeding in dialysis patients may require nephrectomy. Perirenal hematoma following trauma is rare and well identified
Infections:
Infection of the upper urinary tract should be evoked in any febrile PRAD patient.
Frequent in women, it usually follows a cystitis due to enterobacteria, attesting to an ascending extension. Nephritic colic and progressive renal failure associated with infection suggest an associated urinary obstruction: ultrasound and urography are useful exams. Distinguishing kidney parenchyma infection and intracystic infection is difficult: during intracystic infection the urine may remain sterile, whereas fever and local signs suggest infection of the renal lodge.Ultrasonography and CT can help locate the infected cyst by identifying heterogeneous cystic contents or a thick, irregular wall. If uroculture and blood cultures are sterile, ultrasound-guided puncture of a suspect cyst is useful for isolating the causative organism.
The initial antibiotherapy is adapted according to the information emanating from the antibiogram.
However, not all antibiotics diffuse readily into cysts: an appropriate local concentration is achieved quickly only with fluoroquinolones and trimethoprim. This result is obtained after 5 to 7 days of treatment with aminoglycoside or betalactamines. In practice, if the initial gravity is of concern – septicemia or septic shock – empirical parenteral antibiotic therapy with third-generation cephalosporin or fluoroquinolone is indicated, possibly associated with an aminoglycoside. Subsequently, or immediately if the signs of infection are moderate, oral treatment with fluoroquinolone or trimethoprim-sulfamethoxazole is indicated. Apyrexia is sometimes obtained only on day 10-15;percutaneous drainage of an infected cyst may be helpful: except for clinical concern, patience is required. The optimal duration of treatment is not codified: we continue antibiotic therapy 3 weeks in case of parenchymal infection, 6 weeks if it was an intracystic infection.
Calculations:
Their prevalence is high in polycystic kidneys, both in men and women, reaching 11 to 34%: the possibility of lithiasis migration should be considered before any flank pain. About 60% of these lithiases are uric acid and therefore radiolucent, and 40% are made of calcium oxalate.
Several specific factors contribute to increased lithogenesis in PRAD: precalcular canalicular ectasia, observed in 15% of patients; lower urinary pH and hypocitraturia; a urinary stasis linked to the deformations of the excretory cavities by the cysts. On the unprepared abdomen (ASP), distinguishing calcium lithiasis and calcifications from the cystic walls of previous anterior hemorrhages is often difficult: CT scan without, then with contrast injection and intravenous urography (IVU) may be useful in symptomatic patients.
Urinary alkalinization is indicated in case of radiolucent lithiasis; symptomatic oxalocalcic stones can be treated by percutaneous surgery, or extracorporeal lithotripsy.
Kidney Cancers:
Their incidence and the mortality for which they are responsible are identical in PRAD and the general population.Routine screening is therefore not indicated. Cancers associated with PRAD are often multi-center, bilateral, and of a particular histological type, sarcomatous. Fine-section CT is the most cost-effective test for diagnosis. The coexistence of cysts and solid tumors of the kidney necessitates the removal of another hereditary renal disease, von Hippel-Lindau disease.
Hypertension:
Blood pressure elevation is a common and early occurrence in PRAD, which soon has a cardiac counterpart in the form of left ventricular hypertrophy detectable by ultrasound even before HTA or renal function declines. . This fact has been well established in children and adolescents whose renal cysts were still small. It is sometimes on the occasion of a pregnancy HTA that the PRAD is discovered. The prevalence of hypertension is increasing with progression of renal failure, and 80-90% of patients with end-stage renal disease are hypertensive.
The mechanisms generating HTA while the glomerular filtration rate is not yet affected are incompletely elucidated: an early increase in plasma volume contributes, as well as an increase in plasma renin activity inappropriate for hypervolemia. Possibly parenchymal ischemia caused by cystic delivery of the vessels stimulates renin release.Curiously, the presence of cells capable of synthesizing renin has been demonstrated in peri-cystic vessel walls; in addition, the periosteal epithelium seems to be able to secrete renin into the cystic fluid.
Cardiovascular events are the leading cause of death in PRAD patients. The prevention of risk factors, and therefore the early treatment of hypertension, is highly desirable: the goal is to bring blood pressure below 130-140 for systolic, 85-90 for diastolic. A limitation of the intakes of soda at 6 g / d is useful. The use of a product blocking the renin-angiotensin system is initially logical for the physiopathological reasons developed above: beta-blockers or inhibitors of the conversion enzyme (IEC) constitute the first stage of the treatment.
Rare episodes of acute renal failure have been observed during the introduction of ACE inhibitors in patients with chronic renal failure. If HTA is poorly controlled by monotherapy, diuretics are indicated; a moderate dose of furosemide usually remains effective at an advanced stage of renal failure.
Renal insufficiency: progression
Severe renal failure is the most common and serious complication of PRAD. It is not inevitable, and about 20 to 25% of patients reach the age of 70 years free from severe renal failure. For patients whose disease requires renal replacement therapy, dialysis is started on average at age 55: before age 40 in 15% of patients, between 40 and 59 in 75%, and after age 60 in 10 %.
Within the same family, there is no match for age of onset of dialysis, nor anticipation from one generation to the next.The age at which renal impairment begins is variable, but once initiated, the annual decline in glomerular filtration rate is 5 to 6 mL / min.
In a very small number of families, end-stage renal disease occurs in childhood or adolescence: this has only been observed in families whose PRAD is linked to PKD1; the genetic abnormality involved is sometimes caused by a large deletion of PKD1 and a contiguous gene, TSC2. Neomutation in the parent vector or maternal transmission of the disease appear to favor these aggressive forms. In the siblings the risk of recurrence of a serious form is high, and estimated at 43%.
What factors, genetic or otherwise, hasten the progression of kidney failure?
Several concordant studies indicate that end-stage renal disease occurs on average 10 to 15 years later, that is, around age 70 in families not linked to PKD1 (presumed PKD2). Kidney cysts later appear in these patients. End-stage renal disease occurs earlier in patients whose PRAD-free parent has essential hypertension and about 5 years earlier in men than in women. Once renal impairment is established (creatinine clearance less than 55 mL / min), a very rigorous control of blood pressure (mean AP less than 92 mmHg) is not more effective in slowing its progression rate than more usual goal (average PA less than 107 mmHg). The drastic protein restriction (0.3 g / kg / day) or rigorous (0.6 g / kg / day) has no more favorable influence than a conventional restriction (1.2 mg / kg / day) .
Pregnancy:
The majority of PRAD patients’ pregnancies go smoothly, most with normal blood pressure and kidney function. A pregnancy HTA is possible; preeclampsia occurs in 11% of hypertensive patients before pregnancy.
Two studies indicate that pregnancies do not alter the progression of kidney disease, another reports an increased incidence of renal failure beyond three pregnancies if a hypertension is associated. In a young PRAD hypertensive woman, it is essential to postpone a pregnancy project until the good control of the arterial pressure. The fetal prognosis is related to renal failure and control of blood pressure. If marked renal impairment (serum creatinine greater than 200 μmol / L) exists at the time of conception, prematurity and fetal hypotrophy or irreversible impairment of maternal renal function are common, and pregnancy should be discouraged.
Extrarenal manifestations:
Liver complications:
The onset of liver cysts is later than that of renal cysts: in ultrasound, their prevalence increases from the age of 20 to stabilize at 80% around 50-60 years. Anatomically, liver cysts derive from biliary microhamartomas called von Meyenburg complexes: they are biliary tract proliferative foci that have failed to regress during fetal life; hepatic cysts rarely communicate with the bile ducts. Usually these liver cysts are asymptomatic. They are earlier and more numerous in women, where sometimes their size or number is responsible for a polycystic liver disease much more troublesome than polycystic kidney: the repression of neighboring organs by a monstrous liver causes early satiety and nausea, sometimes malnutrition, hernias and respiratory discomfort. Jaundice compression of the bile ducts is rare. Ascites is possible: the compression of the hepatic or inferior vena cava veins in the hepatic segment causes discomfort to the flow of the venous flow which can lead to centrilobular fibrosis; thrombosis of these veins (Budd-Chiari syndrome) has sometimes been observed. Even in these major forms, the hepatic synthesis capacities (factor V or albumin) are rarely altered, the normal liver parenchyma being repressed but persistent. Elevation of alkaline phosphatase is possible.
Asymptomatic polycystic liver disease (PKH) requires no treatment. For very uncomfortable patients, puncture-alcoholization of prominent, bulky but small cysts, or laparoscopic fenestration are useful. For patients whose cystic disease is diffuse hepatic resection is preferable. This liver surgery is dangerous, even in the hands of specialized surgeons, with an estimated mortality of 10%; postoperative ascites is commonplace. Liver transplantation is indicated only in cases of associated hepatic insufficiency.
Acute complications associated with liver cysts include hemorrhage, rupture, torsion, and infection. Fever and pain of the right hypochondrium put on the track of the latter.
The causative organism is usually a Gram-negative bacillus isolated by blood culture or puncture of the infected cyst.The precise identification of the site of infection is arduous and ultrasound, CT scan and MRI can provide additional information, but also underestimate the extent of infection. The treatment combines antibiotherapy and drainage. An antibiotic combination including ciprofloxacin is desirable.
Other hepatobiliary abnormalities are much rarer: idiopathic intra- or extrahepatic dilatation of the main bile duct;congenital liver fibrosis responsible, from childhood, for portal hypertension with splenomegaly and digestive bleeding;cholangiocarcinoma.
Intracerebral aneurysms:
Their prevalence in PRAD is 8%, five times higher than in the general population; it rises to 16% if a related PRAD himself has an aneurysm. The aneurysms are saccular and develop at the Willis polygon bifurcation sites. They are multiple in a third of cases. Aneurysmal rupture is one of the most serious complications of PRAD. It can occur at any age, including before age 20 and in the absence of hypertension. The meningeal haemorrhage is fatal in half of the cases; it may be preceded by localized headaches, unusual in their intensity, which are an alarm signal indicating cracking. Diagnosis is based on cerebral CT and lumbar puncture. Surveillance and treatment are the responsibility of neurosurgery: the early exclusion of the aneurysm by placing a clip on his collar is the traditional solution; if the topography is suitable, selective endovascular occlusion may be preferred. In survivors, new aneurysms may appear.The concentration of cerebral aneurysms in some families suggests an MRI or helical CT screening between 20 and 40-50 years of age to the PRAD relatives of patients who have broken an aneurysm in order to apply preventive treatment if this screening is positive.
Extracerebral cardiovascular abnormalities:
A high prevalence of mitral valve prolapse and various valvulopathies has been reported and recently challenged.These abnormalities rarely become symptomatic. In the absence of heart murmur or rhythm disorder, no monitoring is indicated. The frequency of aneurysms of the aorta is not known. Rare and perhaps fortuitous associations have been detailed in two recent general journals.
Other anomalies:
Cysts can be detected in the pancreas, spleen, esophagus, ovaries, uterus, as well as in the brain (arachnoid cysts).In dialysis patients, the prevalence of colonic diverticula is considered excessive and is associated with an increased incidence of infectious complications. The prevalence of abdominal hernias is also increased.
Genetics:
Genes of PRAD:
The autosomal dominant mode of inheritance of PRAD means that any child of an affected subject is at 50% risk of inheriting the abnormal gene and developing the disease. Two identified genes promote PRAD, PKD1 located on the short arm of chromosome 16 and PKD2 located on the long arm of chromosome 4. In Europe, PKD1 is responsible for 85% of PRAD, and PKD2 for almost all non-related forms at PKD1. Exceptional families are known whose PRAD is not linked to any of these two genes: these facts indicate a heterogeneity of the PRAD. Inter and intra-familial phenotypic heterogeneity of renal disease has been described above. Hepatic cysts and cerebral aneurysms have been observed in families related to PKD1 or PKD2.
PKD1 gene and polycystine:
The PKD1 gene spans 52 kb and has 46 exons. It codes for a protein called polycystin made of 4,302 amino acids. It is a glycoprotein with several transmembrane domains, a large extracellular portion and an intracytoplasmic end. The structure of the polycystine is original and its precise role is not PKD1 is arduous because several fragments of the gene are repeated nearby on the short arm of chromosome 16, which give rise to very close transcripts. Nevertheless, in about 20 PRAD families, the precise nature of the PKD1 mutation has been identified; it is sometimes the production of an abnormal polycystine, sometimes the total absence of protein production is observed. In the latter case, the genetic abnormality involved is a deletion affecting PKD1 and the contiguous TSC2 gene that results in severe polycystic kidney disease in childhood associated with tuberous sclerosis. Studied by immunostaining, polycystine is expressed in the fetal kidney and to a lesser degree in the adult kidney, mainly the tubules; Paradoxically, its expression is intense in cells lining the cyst wall, suggesting that the mutations associated with the disease cause overexpression of the gene.
PKD2 gene:
It encodes a transmembrane protein of 968 amino acids with six transmembrane domains and both ends of which are intracellular. This protein has a partial analogy with a flap of the PKD1 product, as well as with voltage-gated calcium channels. The narrow phenotypic parallelism observed in PKD1 and PKD2 patients suggests that the two proteins interact on a common transduction mechanism, which remains to be identified. Perhaps PKD1 would regulate the activity of a PKD2 channel.
Diagnostic:
Positive diagnosis:
Ultrasonographic evidence of two enlarged kidneys with multiple, contiguous cysts in the cortex and medulla is suggestive of PRAD. Two additional facts contribute to the diagnosis: the presence of liver cysts; a family history characterized by an autosomal dominant inheritance of nephropathy. The diagnosis is more difficult in the absence of family history, in the rare predominantly unilateral presentations, or in young subjects whose cysts are few. We must then make the most of two sources of information.
Ultrasound data:
The minimum criteria for ultrasound diagnosis were specified by Ravine as a function of age in patients with PKD1-related disease. They take into account the facts observed in the general population where renal ultrasound detects at least one renal cyst in 2% of subjects aged 30 to 49 years, 11% of subjects aged 50 to 70 years and 22% beyond. In the PRAD the cysts develop very gradually and usually they are not large enough to be detected by ultrasound in infancy. Nevertheless in some cases they can be identified in the fetal kidney or very early in life. In an at-risk patient from an affected family, the presence of at least two renal cysts (uni- or bilateral) before the age of 30 is sufficient to establish the diagnosis; between 30 and 59 years, the presence of at least two cysts in each kidney is necessary; and in subjects 60 years of age or older, the presence of at least three cysts in each kidney must be required. These criteria take into account that the sensitivity of ultrasound is not excellent for detecting small renal cysts before the age of 20, but that its specificity is strong because solitary renal cysts, a fortiori bilateral, are very rare at this age.Conversely, since renal cysts, which may be bilateral, are relatively common after age 50, stricter criteria must be required to establish the diagnosis at this age; the discovery of multiple hepatic cysts is obviously a strong argument in favor of PRAD.
In practice, the absence of a renal cyst at the age of 30 completely reassures a subject at risk. In families with PKD2-related disease, the onset of cysts is sometimes delayed and the reliability of the ultrasound criteria has not been calibrated to such good accuracy. Finally, it must be recognized that CT scan detects small cysts better, but is not routinely indicated for the diagnosis of PRAD.
Genetic approach:
The precise identification of the PKD1 gene was only established in 1994. Although the first mutations have been reported, the size of the gene and its complexity remain as many obstacles to direct genetic diagnosis. The PKD2 gene has just been identified. For both forms of the disease, genetic linkage analysis remains the practical approach. It should be kept in mind that it requires the study of at least two subjects with the same family, in addition to the proposal, and if possible its unharmed parent. Link analysis is not exploitable if only one subject is reached. The use of multiple genetic markers makes it possible to specify which gene is involved in a given family, and to indicate in about 90% of cases if a test subject is affected. The implementation of the genetic diagnosis is indicated in the subjects at risk who request it, in case of uncertainty at the end of the explorations of imagery, for example with a view to kidney donation if a family transplant is envisaged, or if a pregnancy is planned, or if the search for a cerebral aneurysm is indicated.
Differential diagnosis:
Various rare and hereditary renal cystic diseases can simulate PRAD: their main clinical and genetic features are detailed more
low. Glomerulocystic disease is exceptional; sometimes sporadic, sometimes autosomal dominant, it progresses to renal failure; cysts are small, and developed at the expense of glomeruli. Cystic disease of the renal medulla is observed in adults with autosomal dominant inheritance. The CT scan shows the particular topography of cysts confined to the medulla. Tuberous sclerosis in Bourneville is genetically characterized by autosomal dominant inheritance and a high frequency of neomutations estimated at 50%. Two genes are identified, TSC1 and TSC2; the latter is contiguous with PKD1 and a large deletion affecting both genes gives rise to severe polycystosis from childhood. Renal lesions occur in two-thirds of patients: angiomyolipomas, multiple cysts or possibly associated malignancies.
Liver cysts are possible. Extrarenal manifestations include convulsions, mental retardation and periventricular calcifications; fibroids of the face and peri-inguinal regions; cardiac rhabdomyoma and pulmonary lymphangiomyomatosis.
Von Hippel-Lindau disease is also autosomal dominant. It gives rise in the kidney to cysts and clear cell cancers.These lesions are bilateral and multiple. Elsewhere there are hemangioblastomas of the retina or central nervous system, pheochromocytomas and cysts of the pancreas. Orofaciodigital syndrome type I is transmitted on the X-linked dominant mode. Lethal in male fetuses, it gives rise to a bifurcated tongue and palate, digital abnormalities and mental retardation.
Among the non-hereditary cystic disorders, some are congenital and unilateral: multi-cystic dysplasia of the child, where the kidney is mute; multilocular cysts where CT shows intracystic walls. Others are acquired: parapyelic cysts;cysts associated with renal failure of chronic nephropathies or dialysis: the kidneys are then small with cysts protruding largely at their periphery.
Antenatal diagnosis:
An antenatal diagnosis by genetic linkage study on fragments of chorion taken at the ninth week of pregnancy can be done at the request of the parents if the family is informative – that is to say that the gene in question was previously identified – and that parents want a termination of pregnancy in case of a positive response. An interview with a geneticist is essential. Experience shows that the desire to terminate a pregnancy is in fact very rare in PRAD families, perhaps because the disease is rarely serious before the sixth decade.
Pathophysiology:
While waiting for precise information on the function of PKD1 and PKD2-encoded proteins, it is important to stick to the mechanisms of kystogenesis. The growth of a cyst requires the proliferation of the border cells and the secretion of the cystic fluid. The renal cysts of PRAD develop from any segment of the nephron, from the Bowman capsule to the collecting tube. Only 1 to 2% of nephrons are affected by this cystic transformation. The cause of this disparity is unknown. By growing the cyst loses its connections with the normal tubule. The cystic fluid is secreted by active mechanisms. Its concentration of sodium and protons varies, probably according to the tubular segment from which the cyst is derived (proximal tubule: concentrations identical to those of the plasma, distal tubule: low concentration of sodium, high in protons).
The cystic fluid is also rich in amino acids, TNF (tumor necrosis factor) alpha, EGF (epithelial growth factor), IL1 (interleukin 1), renin and erythropoietin. The secretion rate of the fluid is increased in vitro by cyclic AMP (adenoside monophosphoric acid) and a secretagogue present in the cystic fluid itself.
Various membrane proteins are poorly located in the cystic epithelium: for example the EGF receptor, ankyrin and fodrin are at the apical pole; the Na + -K + -2Cl – transporter is at the basal pole; Na-K-ATPase is at the apical pole and has increased activity. Although these latter facts have been contested by others, they suggest an anomaly in the routing of various proteins in PRAD.
Mechanism of proliferation of cystic cells:
Different facts can be enumerated: the cystic epithelium has an increased sensitivity to EGF whose concentration is high in the fluid; inadequate expression of transcription factors such as Pax 2 and WT-1 is established.
In an experimental model, disabling the bcl-2 gene, which normally inhibits apoptosis – that is, programmed cell death – results in polycystic kidneys; in another, the use of taxol, which interacts with microtubules, inhibits the formation of hereditary renal cystic disease. In the kidneys of PRAD patients, the proportion of apoptotic cells is excessive, both in the cells lining the cysts, in the non-cystic epithelia, and in the interstitium, whereas the apoptosis is almost not detectable in the cells. normal kidney.
Interactions between epithelial cells and interstitium play a key role in cell ontogeny and polarization, and may be altered by mutations affecting polycystin or its interactions with the PKD2 gene product. It is perhaps in the understanding of these mechanisms of kystogenesis that the possibility of targeted pharmacological interventions lies.
Finally, the histological lesions of the polycystic kidney include a marked interstitial fibrosis, possibly favored by the molecular anomalies that have just been reported.
Treatment of end-stage renal failure:
Hemodialysis and renal transplantation can be proposed, overall survival is comparable regardless of the approach used and averages 14 years. It is therefore identical to or slightly greater than that observed in patients receiving hemodialysis for another variety of primary nephropathy; graft survival is identical. The transplantation of a kidney from a related donor requires a total exclusion of the possibility of PRAD in the donor, by renal CT or genetic linkage study.Cystic disease does not relapse on the transplanted kidney.
Before transplantation, it is advisable to perform a uni- or bilateral nephrectomy if available for exchange and compromising the respiratory tolerance of abdominal repletion.
In dialysis or transplanted PRAD patients, cardiovascular mortality is high, similar to what is observed with these methods of treatment. The prevalence of stroke is a little increased. Infections of the kidneys or liver cysts are common.
ALPORT SYNDROME:
Alport Syndrome (AS) is an inherited renal disease characterized by the occurrence in several generations of the same family of progressive hematuric nephropathy and perceptive deafness. Renal involvement, more severe in men is responsible for 1 to 2% of terminal renal failure. It is due to changes in the basement glomerular membrane, resulting from genetic abnormalities affecting the chains of collagen IV, the main component of basement membranes.Transmission occurs in 85-90% of the families according to the X-chromosome dominant mode, but in the other families the mode of transmission is autosomal, recessive or dominant.
Clinical manifestations of Alport syndrome related to X:
Nephropathy:
Microscopic hematuria can be observed in the first days of life or in childhood. This microhematology allows early diagnosis of the condition since it is detected in all children affected. Episodes of macroscopic haematuria occur in 50% of cases, most often before the age of 5; they gladly succumb to infection of the upper respiratory tract. Their frequency decreases with age and they become exceptional after 15 years. The incidence of proteinuria increases with age: often absent in childhood, it becomes minimal and intermittent and permanent especially in affected boys. In 40% of cases, moderate nephrotic syndrome is seen after the age of 10 years. This nephrotic syndrome of poor prognosis may be accompanied by hypertension. The evolution of renal failure is heterogeneous; it depends on the sex of the patient and other genetic factors. In all male patients, progression to end-stage renal disease (ESRD) is inevitable: it rarely occurs before the age of 10, often between the ages of 15 and 30 years. In these severe forms, the intrafamilial homogeneity of the age of occurrence of the IRT is striking. In a smaller group of families, the IRT is observed at 40 years on average with extremes ranging between 30 and 70 years, and a clear intrafamilial heterogeneity. In the vast majority of affected women, intermittent or permanent microscopic hematuria is detected, with or without associated proteinuria.
Only 5 to 15% of women with the disease progress to the IRT at a slow pace. The intensity of proteinuria and renal histological abnormalities provide predictive indications; the proportion of renal cells whose normal X chromosome has followed random inactivation may account for the phenotypic heterogeneity of X-related diseases in women; the nature of the mutation affecting type IV collagen could also contribute to the phenotype.
It appears in half of the cases in the boy before the age of 10 but can remain latent and be detected only in the audiogram which is indicated in any suspect subject of SA.
Hypoacusis can progress in childhood to require an apparatus. This fact is rarer in adulthood and exceptional in women. The intensity of hearing loss does not prejudge the course of nephropathy. At the extreme end of the spectrum, families with SA-related nephropathy and COL4A5 gene defects (encoding the α5 collagen IV chain) that determine collagen IV abnormalities are free of auditory abnormalities.
It has been described after renal transplantation a reduction in neurosensory deficit attributed to a reduction in the neurological consequences of chronic uremia.
Eye abnormalities:
Ocular abnormalities mainly related to the lens and retina are found in 15 to 40% of patients with AS. Among the disorders of the lens, the bilateral anterior lenticone is the most specific anomaly, and most often associated with a perception deafness. The diagnosis is based on a biomicroscopic examination which shows the conical protrusion of the lens in the anterior chamber of the eye. The lenticone may be accompanied by an axial myopia of progressive evolution. Opacities of the crystalline lens (cataract) are rather commonplace, but not specific. Retinal lesions are more frequent and mainly consist of pale yellow puddles of the macula or nearby areas. These retinal abnormalities do not result in a decrease in visual acuity or abnormality of the electroretinogram. Finally, frequently neglected corneal erosions have been described.
Diffuse leomyomatosis:
This condition may be sporadic or hereditary and has been reported in association with X-linked AS in 35 patients from 19 families. It is a tumor related to the benign proliferation of smooth muscle cells of the muscular layer of the esophagus – revealed by dysphagia whose treatment is surgical – of the tracheobronchial tree or genital tract in the women (uterine myoma, hypertrophy of the clitoris and vulva). The renal phenotype is severe in boys, bilateral congenital cataract is common.
Biochemical, histological and genetic aspects:
Biochemistry of basement membranes:
The initial abnormality of X-linked AS affects the α5 chain of collagen IV as a consequence of a corresponding gene defect (COL4A5). The major component of the glomerular basement membrane (MBG) is the network-type IV collagen that binds laminin, entactin and proteoglycans. Each type IV collagen monomer is formed of three α chains wound in triple helix. There are six varieties of α chains (designated α1 to α6) and a large number of isoforms of type IV collagen. Each α chain is formed of three segments: the main, median domain is said to be collagenous; at both ends, two non-collagenous domains of different sizes associate in homodimers (at the carboxy terminal end, stabilized by disulfide bridges forming a globular structure and called NC1 for non-collagenic 1) or homotetramers at the amino-terminal end. Two varieties of collagen networks are identified, one labeled (α1) 2-α2, the other formed by α3 and α5 chains. The distribution of the chains α1 (IV) and α2 (IV) is ubiquitous, that of the other chains is restricted. In the kidney, the former are synthesized by the mesangial and endothelial cells and contribute to the basal membranes of the vessels and tubules, as well as to the mesangial matrix and the subendothelial layer of the MBG. The α3 to α5 chains would be formed in the epithelial cells and contribute only to the basement membranes of the lungs. The α5 and α6 (IV) chains contribute to the basal epidermis.
The genes encoding the six α-chains contributing to type IV collagen are paired upside-down on three distinct chromosomes: the COL4A1 and COL4A2 genes are located on chromosome 13, COL4A3 and COL4A4 on chromosome 2 and COL4A5 and COL4A6 on the arm The carboxy-terminal region of α3 (IV) contains the antigenic pattern recognized by the circulating antibodies of Goodpasture’s syndrome.
Morphological study:
The study in light microscopy can show a normal renal parenchyma in the small child. Subsequently, non-specific glomerular lesions appear: enlargement and hypercellularity of the mesangial axes, segmental thickening of the capillary wall, and segmental hyalinosis of the floculus. Early lesions of interstitial fibrosis and tubular atrophy are commonplace; the finding of masses of lipophages passes for suggestive of SA, but can be observed in any nephropathy with abundant and durable proteinuria.
The study in electron microscopy is essential to visualize the most characteristic lesions. The glomerular basement membrane (MBG) appears irregularly thickened with lamination and fragmentation of the lamina densa enclosing clear areas where microgranulations are visible. The outer side of the MBG is scalloped and lined with enlarged podocytes.The thickening of MBG is often diffuse in adults and segmental in children where alternation with areas of thinning can be observed. In the extreme, only a diffuse thinning of the MBG is possible. All these ultrastructural lesions and the absence of immunoglobulin deposits or complement immunofluorescence are strongly suggestive of an SA, and therefore particularly useful to establish a diagnosis in the absence of family history, lack of investigation precise or because of de novo mutation.
Immunofluorescence is usually negative but segmental deposits of IgM or C3 are sometimes present in hyalinosis lesions.
Immunohistochemical study:
It has long been known that the application of monoclonal antibodies directed against MBG but of unknown specificity was oddly associated with no binding in the majority of male patients with AS, opposing the continuous fixation seen in patients with AS. normal subjects; in the affected women, discontinuous or linear fixation was observed. The use of α-chain specific antibodies confirmed and clarified these facts:
Anti α5 (IV) antibodies do not bind in 75% of patients; Notably, there is no binding of anti α3 (IV) or anti α4 (IV) antibodies in an identical proportion of patients. The explanation advanced on this subject suggests that the COL4A5 gene defect (involved in the X-linked SA) would disturb the molecular assembly of the triple helices including the α3 and α4 chains whose formation or stability requires an α5 chain normal.
Genetics:
In about 85% of cases, the SA is transmitted in the X-dominant mode. It is linked to an attack of the COL4A5 gene, localized in Xq22, coding for the α5 chain of collagen IV. The study of more than 150 rearrangements inside the COL4A5 gene showed
that deletions are present in 5 to 15% of families with severe kidney phenotype, associated with hearing loss and ocular abnormalities in 50% of cases. In the majority of families, these are point mutations of the COL4A5 gene: point mutations of the collagen and NC1 domains, mutations at the splice sites, and insertions were observed, each family having a particular mutation. The consequences on the structure of the protein are variable (α5 chains present but abnormal or truncated or even absent) and can induce severe or slow phenotypes with or without deafness.
In patients with AS and diffuse oesophageal leiomyomatosis, the deletions involved carry the first exons of the COL4A5 gene but also of COL4A6 and the intergenic region which contains common regulatory genes.
Alport syndrome with autosomal transmission:
In 10 to 15% of cases, AS is transmitted in the autosomal recessive mode. The suggestive facts of such a transmission are a consanguineous marriage, an identical gravity in both sexes, and moreover, the occurrence of a renal insufficiency before the age of 20 years in a woman. Hearing and eye damage are comparable to what is observed in X-linked AS, as are ultrastructural lesions of MBG. Immunohistochemical counterparts have been recently specified and conclude that heterogeneity is comparable to that observed in X-linked SA. The disease is linked to the COL4A3 or COL4A4 genes located on chromosome 2 and encoding the α3 and α4 chains of collagen IV.
An autosomal dominant mode of transmission affects 5% of families. The most suggestive clinical fact is a transmission of the affection of a father to his son. The genetic anomaly in question is still unknown.Macrothrombocytopenia is possible and has been reported in about ten families, associating thrombocytopenia (between 5,000 and 100,000 / mm3) and the presence of giant platelets with or without abnormal structure or function.Thrombocytopenia can prolong bleeding time and cause bleeding accidents. These platelet abnormalities are usually associated with severe renal impairment leading to IRT before age 25, and pronounced hypoacusis in women.
Treatment:
There is currently no specific pharmacological or genetic treatment that can alter the course of the disease. End-stage renal failure requires extrarenal cleansing or transplantation.
The evolution after renal transplantation is favorable in the majority of the cases, the abnormality of structure of the MBG sparing the transplanted kidney. Nevertheless about 15% of the transplanted patients develop in the year following the transplantation a linear fixation of IgG along the MBG of the transplant. In 2 to 5% of cases a true rapidly progressive extracapillary glomerulonephritis occurs which can lead to the loss of the graft despite the practice of plasma exchanges. The diagnosis is based on the detection of circulating anti-MBG antibodies and biopsy of the graft.This complication is observed especially in humans during X-linked SA and responsible for an early form of the disease. Recurrence is common after retransplantation. The alloimmunization developed against the transplanted kidney is attributed to the introduction of epitopes absent in the native kidneys.
Is it possible to plan a transplant from a related female donor? In so-called informative families, one should not hesitate to use molecular biology to clarify the status of the potential donor. For the others, a histological examination is necessary renal: it is better not to practice a levy likely to harm the donor.
Differential diagnosis:
It is the discussion of a micro- or macroscopic hematuria in the child. Family history, perceptual hearing loss, specific ocular abnormalities, ultrastructural features, the lack of antigenicity of MBG, and even better the demonstration of a COL4A5 gene mutation are the most useful elements for diagnosis. IgA nephropathy and benign familial hematuria should be ruled out. In the latter case, nephropathy does not progress to renal failure, there is no gross hematuria nor proteinuria nor auditory disturbance and basement membranes are usually thinned.
Genetic counseling:
Characterizing the mode of transmission is based on the establishment of a family tree and the bioclinical study of relatives at risk. The identification of female carriers, the possibility of de novo mutation and compliance with diagnostic criteria are serious obstacles. Molecular genetics, by linkage study (informative in 75% of families whose SA is linked to the X) or direct characterization of the mutation (50% of families tested) can be useful. Genetic counseling must be provided jointly by geneticist and nephrologist.
CONGENITAL NEPHROTIC SYNDROME:
Strictly speaking, the term “congenital nephrotic syndrome” applies to cases where this syndrome is present from birth.In fact, the term has been extended to nephrotic syndromes that appeared in the first 3 months of life; many congenital nephrotic syndromes, but not all, are of genetic origin. On the contrary, the term “infantile nephrotic syndrome” is reserved for cases where this syndrome manifests itself during the first year of life and includes, in particular, many cases of nephrotic syndrome with minimal glomerular lesions.
Finnish-type nephrotic syndrome:
It is the prototype of hereditary congenital forms: its frequency is high in Finland (about one in every 10,000 births), but affected families have been identified in other countries of the world, including France. The disease is transmitted according to the autosomal recessive mode; the gene has been located on the long arm of chromosome 19 but has not yet been identified. Affected newborns are often born prematurely and are of small weight. The placenta is usually overweight. Nephrotic syndrome is associated with anasarca, thromboembolic and infectious complications, undernutrition and severe growth retardation.
The renal biopsy revealed lesions of which none is pathognomonic: pedicel fusion, proliferation of podocytes, glomerulosclerosis, increase of the mesangial matrix, microcystic dilation of the proximal tubes, finally negativity of the immunofluorescence examination. The pathogenesis of the disease is unknown.
The treatment upset the prognosis of the Finnish-type nephrotic syndrome; previously, all affected children died before the age of four. The symptomatic treatment must be energetic to correct the consequences, including nutritional consequences, of the nephrotic syndrome, using a gastric tube and a venous access route. The next steps include “medical” or surgical biphrectomy, chronic peritoneal dialysis and kidney transplantation. In about 25% of cases, nephrotic syndrome recurrence after renal transplantation.
The nephrotic syndrome sets in during fetal life, resulting in an increase in the concentration of α-fetoprotein in the amniotic fluid. This dosage can be used for prenatal diagnosis in affected families.
Diffuse mesangial glomerular sclerosis:
mutations of the Wilms tumor suppressor gene, WT1, have been demonstrated; most often, the disease is sporadic.Galloway-Mowat syndrome is the second entity where glomerular damage, microcephaly and developmental delay coexist; this syndrome is transmitted according to the autosomal recessive mode.
Segmental and focal glomerulosclerosis:
It is usually seen in familial corticosteroid-resistant nephrotic syndromes that may be congenital but most often develop later and progressively in childhood.
HINDICITED MALIGNANT TUMORS OF THE KIDNEY:
Hereditary forms of nephroblastoma (or Wilms tumors) are rare (2 to 3% of cases); tumors are often bilateral. They can be isolated or associated with various malformation syndromes (Wilms tumor-associated WAGR, aniridia, genitourinary anomalies, mental retardation, WT1 gene in 11p13) and Beckwith-Wiedemann syndrome characterized by gigantism, macroglossia, omphalocele and various malignancies, WT2 gene at 11p15). There is still a third locus also located on the short arm of chromosome 11.
Hereditary renal carcinoma:
Renal carcinoma is hereditary in about 4% of cases. In three families with clear cell kidney cancer, a chromosomal translocation was identified, involving the short arm of chromosome 3, and locating the responsible gene in 3p14.2. In von Hippel-Lindau disease (VHL), clear, multifocal and often bilateral renal cell carcinoma is one of the main manifestations of the disease; the gene is identified and located in 3p25-26. In tuberous sclerosis, a few cases of renal carcinoma have been reported; this inherited disease predisposes to renal cell carcinoma but with a much more moderate prevalence than during VHL. Finally, there are several families with multiple generations of hereditary renal papillary cancer, often multiple and bilateral. The causative gene is located at chromosome 7. Sporadic papillary kidney cancer accounts for 5 to 10% of renal tumors.
REESSIVE RENAL AUTOSOMIC POLYKYSTOSIS:
Autosomal recessive polycystic kidney disease is a rare disease with a prevalence of approximately 1 in 40,000 individuals. The gene was located at chromosome 6p.
Renal disease usually occurs in childhood, is characterized by dilatation of the collecting ducts and is consistently associated with liver injury, congenital liver fibrosis.
In a poorly defined percentage of cases, polycystic kidney disease is marked at birth by huge kidneys and is complicated by pulmonary hypoplasia resulting in fatal respiratory distress in the neonatal period. Often these cases have been accompanied by severe oligoamnios during fetal life.
In other cases, polycystic kidney disease is found on palpation of two large kidneys, or before an infection of the urinary tract, or arterial hypertension. Ultrasound shows the increased volume of kidneys whose echogenicity is increased; cysts are detected at birth in 75% of cases or develop later. The IVU is no longer necessary today for diagnosis: it showed two large kidneys with streaks or flamed aspects drawing the dilated collecting ducts.
In children who do not die in the neonatal period, kidney failure develops, often more slowly than previously thought;this is due to the quality of the antihypertensive treatment because the hypertension is often severe in childhood, but can curiously improve thereafter. About one-third of the affected patients progress towards end-stage renal failure in childhood; the others do it more slowly, beyond the age of 15 or even 20 years.
Congenital liver fibrosis (or biliary dysgenesis) is characterized by portal fibrosis and proliferation of intrahepatic bile ducts. It is manifested by hepatomegaly and gastrointestinal bleeding by portal hypertension, requiring medical treatment and sometimes a portocaval anastomosis.
There is no liver failure. Intrahepatic bile duct dilations (Caroli syndrome) may be associated with episodes of cholangitis. In 30 to 50% of cases, hepatic involvement is asymptomatic, detected only by abdominal ultrasound showing discrete signs of portal hypertension or intrahepatic biliary dilatation. Liver biopsy may be useful in some cases.
The diagnosis of polycystic recessive disease is based on ultrasonographic, renal and hepatic data, and on the absence of renal cysts in sonography in both parents, excluding polycystic dominant disease (but not excluding a very rare neomutation associated with polycystic dominant early manifestations). The demonstration of congenital liver fibrosis is decisive in these latter cases. Polycystic recessive disease can be observed exceptionally in adults over 30-40 years of age: liver damage is symptomatic, there is no liver cyst, kidney disease causes chronic renal failure.
NEPHRONOPHTISE AND RELATED DISORDERS:
Juvenile nephronophthis is an autosomal recessive disorder, leading to end-stage renal failure by the age of 20; at this advanced stage medullary cysts or located at the corticomedullary junction are highlighted.
This is why nephronophthia is often associated with cystic disease of the renal medulla, a disease observed in adults and transmitted in the autosomal dominant mode. Many clinical and genetic features differentiate these two diseases;Molecular genetics and its consequences will tell us in the future why these two diseases share cysts of the renal medulla.
Nephronophthis accounts for about 10% of the causes of end-stage renal failure (ESRD) in children; Among the inherited diseases evolving towards the IRT at this age, the nephronophtise is in first place (approximately a third of the cases), well before the polycystose recessive, the syndrome of Alport, the congenital nephrotic syndrome etc. The disease is revealed, between 4 and 6 years, by polyuria-polydipsia and secondary enuresis, witnesses of the lack of concentration of urine, by episodes of extracellular dehydration due to urinary leakage of NaCl, a pallor due to anemia, often more marked than in other nephropathies, fatigue, pruritus or stunting. HTA is rare. Proteinuria is weak or absent; the urinary sediment is normal. End-stage renal disease is reached at an average age of 11 to 13 years (between 4 and 20 years). Kidney biopsy shows chronic tubulointerstitial lesions; the most striking, but not pathognomonic, anomaly is the thickening and puffing of the tubular basement membrane with abrupt transitions between very thickened segments and thinner areas. It is not known today whether or not the primary lesion touches the tubular basement membrane. Medullary cysts are usually detectable only at an advanced stage.
The juvenile nephronophtis gene was localized by Antignac et al on the short arm of chromosome 2, in the 2q13 region. Large homozygous deletions were found in 85% of affected children; the study in molecular genetics is of diagnostic interest since a comparable deletion is found in 65% of “sporadic” cases (which belong to the same entity).
The association of juvenile nephronophtis with retinitis pigmentosa is known as Senior-Loken syndrome (about 12% of the total). Early-onset forms are similar to Leber’s congenital amaurosis, with blindness at birth or in the first 2 years.On the contrary, in later forms, bilateral tapetoretinal degeneration leads to blindness at school age. Other eye abnormalities may be associated. Molecular genetics have shown that Senior-Loken syndrome is not related to the locus of juvenile nephronophthis; it is therefore an independent disease (even if the renal lesions are identical in both entities). It is worth remembering that the association of nephropathy-retinitis pigmentosa is found in other hereditary diseases, in particular Bardet-Biedl syndrome.
Other extrarenal manifestations are more rarely associated with nephronophthisis: hepatic fibrosis, cerebellar ataxia, bone abnormalities with epiphyses cones.
The treatment of nephronophthis is symptomatic. The disease does not recur in renal transplantation.
Cystic disease of the renal medulla is seen in adults, progressing to end-stage renal failure between about 25 and 40 years of age. Cysts can be identified earlier by imaging. Renal histopathological lesions resemble those observed in juvenile nephronophthis. On the other hand, there are no associated extrarenal manifestations, and in particular no retinal involvement. The mode of transmission appears to be autosomal dominant in most cases.
PRIMITIVE HYPEROXALURIA:
Two main varieties of hereditary primary hyperoxaluria (autosomal recessive) are described: type I and type II.
Primary hyperoxaluria type I:
It is due to an abnormal metabolism of glyoxylate that is not properly metabolized to glycine due to the defect of a urinary peroxisomal enzyme are composed, remarkably, of calcium oxalate monohydrate).
AGT uses pyridoxine (or vitamin B 6 ) as a coenzyme. In patients, the AGT activity measured on a liver biopsy fragment is lowered; in some cases, AGT is poorly located in the cell, present in mitochondria and absent or rare in peroxisomes. Finally, the gene coding for AGT has been identified and located on the long arm of chromosome 2;mutations have been detected in affected families.
The disease can be very early in life with massive nephrocalcinosis leading to progressive renal failure. Often the disease occurs later in childhood, between 2 and 18 years, marked by recurrent oxalocalcic urolithiasis, rapidly associated with nephrocalcinosis.
Renal function changes more or less rapidly, sometimes during a surgical procedure or a urinary obstruction by a calculation. As kidney failure worsens, calcium oxalate is deposited in many tissues, resulting in oxalosis: arterial walls (explaining finger gangrene), bones and joints, causing pain and impotence, myocardium (resulting in atrioventricular block), nerves and retina in particular.
The same is true of many dialyzed children and young adults with primary hyperoxaluria because oxalate production usually exceeds the dialysis clearance capacity. This is why dialysis and transplantation must be considered early, before the body accumulates too much calcium oxalate.
Finally the disease can only be revealed in adulthood, with a slower destruction of the kidneys, between 30 and 60 years. The rate of progression is not directly related to the intensity of the enzyme deficiency.
The diagnosis of type I hyperoxaluria is based on the following elements (in addition to clinical and possibly familial history): the identification of calcium oxalate crystals and stones monohydrate, the massive urinary excretion of oxalate and glycolate , and – especially in the anuric patients – the enzymatic deficit established by puncture-liver biopsy.
The treatment involves the following steps: ingestion of at least 3 L / d of water and possibly therapeutic measures to promote the urinary inhibitors of crystallization (Mg, citrate or reduce calciuria (thiazide); partially or almost completely sensitive to the administration of pyridoxine, by dosing the oxaluria before and after vitamin B 6 , to carry out the dialysis early by seeking an optimal purification of the oxalate, to quickly prepare the recourse to the transplantation: at the child, often combined transplantation of the kidney and the liver (which makes it possible to correct the enzymatic deficiency) or sometimes isolated renal transplantation in the adult, provided that special precautions are taken after the transplant to ensure adequate purification of the oxalate accumulated calcium in tissues, or even isolated liver transplantation in some rare cases in young children.
Hyperoxaluria type II:
It is due to a defect in D-glyceride dehydrogenase (which can be assayed in leukocytes). The disease appears much rarer and milder than type I hyperoxaluria, although some cases of renal failure have been described.
CYSTINOSIS:
Cystinosis is a rare autosomal recessive inherited metabolic disorder (1 in 20 000 to 300 000) characterized by intracellular accumulation of cystine in lysosomes (due to a defect in the transport system that expels cystine lysosomes). The gene was localized to the short arm of chromosome 17. Only cystinosis with renal involvement will be considered here; the form without nephropathy includes only corneal deposits.
The disease is most often found between 3 and 18 months by digestive disorders, fever and stunting. It is characterized first by severe Fanconi syndrome and its consequences, then by progressive renal failure, resulting in the terminal stage in 50% of cases around the age of 9 years. Renal involvement may, however, occur later at 4-5 years, in adolescence, or even adulthood. The diagnosis is based on tissue accumulation of cystine and the simplest test is the determination of cystine in leukocytes.
Extrarenal manifestations are the result of this accumulation in other tissues of the body and this continues even after dialysis or kidney transplantation: growth retardation and pubertal delay, deposit in the cornea and retina threatening vision, hypothyroidism, hypogonadism, diabetes mellitus by pancreatic involvement, progressive brain damage.
Symptomatic treatment is aimed at correcting the disorders caused by Fanconi syndrome, extrarenal disorders and renal failure, including dialysis and renal transplantation. The accumulation of cystine is reproduced in the transplant, usually without major clinical consequences.
In fact the major therapeutic progress is represented by cysteamine, an aminothiol that enters the lysosomes, forms a cysteine-cysteamine complex that can leave the lysosome and thus reduce the cystine overload.
Oral administration of cysteamine and its analogues, early (before the age of 2 years) and in sufficient doses (provided that tolerance and adherence are adequate), slows or prevents progression to renal failure ( without modifying the Fanconi syndrome). Cystemic eye drops also reduce corneal deposits of cystine crystals.