 Introduction:
Introduction:
The idea was expressed at the end of the 19th century that hypertension could lead to renal sclerosis, and it was Volhard and Fahr in 1914 who introduced the term “hypertensive nephrosclerosis”. This concept remains discussed. While it is perfectly clear that malignant hypertension can lead to end-stage renal failure, the idea that benign hypertension may lead to the same result is far from being accepted by all.
It is traditional to say that the kidney is both guilty and a victim of hypertension. The “guytonian” approach to hypertension was accepted in the 1970s.
Not only did no one come to contradict her, but the evidence accumulated and grew stronger over time. In this approach, no hypertension can be permanent if the renal function is normal, and not obeyed by an anomaly of the equilibration of the sodium balance (function devolves to the only kidney). In this sense, the kidney is indeed the fundamental “culprit”.
The opposite view is that renal physiology can not remain indifferent to a high level of pressure. Renal hemodynamics is rapidly modified, in fact especially in quite severe hypertension. Over time, these changes are no longer only functional and appear anatomical lesions, called “nephroangiosclerosis”. Long-term renal failure occurs, more or less severe, but sometimes reaching the end stage. This problem is considered a rarity, even a curiosity for nephrologists in our country. On the contrary, it is a very tangible reality, common in the United States among the African-American population.
It is therefore a vicious circle that is engaged between the kidney and the hypertensive disease. Like any vicious circle, this one raises the question of “the chicken and the egg” and the idea that a nephropathy can precede and be responsible for hypertension is defended by a growing number of authors.
Definitions:
Two rather different affections are listed under this term.
The most common, described here, is “benign” nephroangiosclerosis. It is progressive kidney disease, a late consequence of prolonged hypertension, which is usually not or poorly treated. Its anatomical lesions are vascular, sclerous, resulting in late glomerular obsolescence, then interstitial fibrosis. It is progressively evolving towards renal failure. End-stage renal failure is mostly seen in black American subjects. In Caucasian populations renal failure is late, less progressive, and it is rather rare that it progresses to the terminal stage. This is in relative discrepancy with the data on the dialysis registers as shown below.
Completely different is “malignant” nephroangiosclerosis.
It occurs suddenly, in the noisy setting of “malignant” or “accelerated” hypertension, characterized by very high blood pressure figures, a rich symptomatic procession, major alterations of the fundus, sometimes acute left cardiac failure.Renal failure evolves very rapidly in a few weeks to the final stage, it is most often irreversible. Its anatomical lesions are different from those observed in the chronic form, marked by acute vascular and glomerular lesions consisting of fibrinoid necrosis and thromboses. This type of nephroangiosclerosis has become very rare nowadays. We will not consider it in detail here.
Frequency. Epidemiology:
There are no truly reliable epidemiological data. It is indeed considered “nephroangiosclerosis” any chronic renal failure with no known cause other than hypertension. Data from dialysis registers provide an approximation of the estimated frequency, at least estimated, of nephroangiosclerosis, but biased by the uncertainty of diagnosis. Data from kidney biopsies provide much more rigorous information, but their limit is that of the relatively selective indications of this examination.
Dialysis registers:
The incidence of hypertension-related end-stage renal disease in the United States is 1 in 2 200 hypertensive patients.It is stable or in very slight progression for ten years.
Data from the US Renal Data System shows 63,800 patients who have dialysis with hypertension as the presumed cause of their kidney disease, accounting for 25% of the dialysis population, making it the second leading cause after diabetes. In incidence, hypertension would justify 29% of new cases of dialysis. It should be noted that hypertension is the presumptive diagnosis in 36.8% of black Americans compared to 26% in white subjects. The frequency would be slightly lower in Latin America, 21 to 22%. Data from the European Register of the European Dialysis and Transplant Association (EDTA) (not including France) indicate a significantly lower annual incidence, accounting for 12% of dialysis treatments. The frequency would seem even lower in Asia, with rates of 6% for Japan and 7% for China.
It should be noted, by contrast, that this rate is 24% for Asian patients dialysed in the United States.
Some authors have pointed out that antihypertensive treatment has significantly improved the cardiovascular outcome of hypertensive patients, perhaps allowing more of them to reach an advanced stage of their renal disease. Moreover, all these data are very sensitive to the age limit of dialysis in the regions considered.
Kidney biopsy data:
Any different is the first provided by kidney biopsy studies. The Norwegian Kidney Register contains data for all renal biopsies performed in this country. Over a 2-year period (1988-1990), 1,176 renal biopsies were performed, and 102 found nephroangiosclerosis.
Biopsies showing any other form of nephropathy, including diabetic, were excluded. Thus, 8.7% of all renal biopsies performed in this country over a 2-year period showed nephroangiosclerosis. Other biopsic data, but lacking this completeness, have been reported. In Denmark, nephroangiosclerosis represents 2.1% of biopsies, and in the United Kingdom 2.5%.
Nevertheless, it remains to be seen which patients are biopsied and on what criteria, because a weighting is very probably necessary.
Clinical picture:
Nephroangiosclerosis is a pathology of remarkable clinical discretion, completely asymptomatic for many years in most patients. A gradual loss of concentration power of the urine can occur, leading to nocturia, rarely important or very troublesome. It is almost always a discovery of systematic monitoring of renal function. Contrary to what is observed in malignant nephroangiosclerosis, there are often no major lesions in the fundus.
Biologically, nephroangiosclerosis presents as a progressive renal failure, very slowly progressive.
Proteinuria is weak, sometimes even nil, and the urinary sediment is almost always normal. In radiography or ultrasound, the kidneys appear of normal size (at least initially) or a little reduced, they are well symmetrical and of perfectly harmonious outline. Doppler arterial shows only symmetrical damping of the flows. It is therefore a fairly naked renal failure, without the urinary syndrome of glomerular nephropathies, and without the abnormalities of the outline of the kidneys observed in many interstitial nephropathies. An angiography is hardly practiced nowadays, except for particular diagnostic hesitation. It shows an impoverishment of the finest arterial ramifications of the cortex, which appear tortuous, irregular, and rarefied.
In the above-mentioned study of the Norwegian registry, patients were 55 years old on average, had a plasma creatinine of 165 μmol and a proteinuria of 0.35 g 24 h -1 , renal disease was known for 1 year . Patient follow-up was 12 years (0.1 to 13.6). End-stage renal failure occurred in 26.4% of patients during this average follow-up period. Age, initial creatinine and proteinuria were the main predictors of this evolution.
Microalbuminuria is increasingly being monitored in hypertensives. Although it appears very clearly as a marker of a very increased cardiovascular risk, it has not so far been very convincingly documented as a predictor of nephroangiosclerosis.
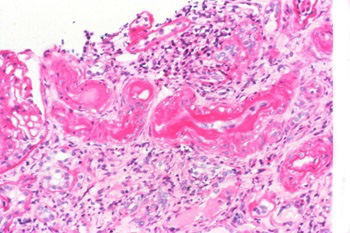
Pathological anatomy:
The histological lesions of nephroangiosclerosis are relatively unspecific. However, if there is no truly characteristic picture, renal biopsy is at least of major interest in eliminating nephropathy of another type.
The histological lesions are essentially arterial and arteriolar. At the level of the interlobular arteries, there is a thickening of the intima, infiltrated by a dense collagen tissue, periodic acid Schiff (PAS) positive, and a multiplication of the elastic blades, narrowing the light. The media is often thickened.
The arterioles, mainly afferent, are the seat of sub-endothelial hyaline deposits more or less extensive, sometimes hyperplasia of smooth muscle cells, and various degrees of sclerosis. The larger arteries are the seat of a variable atheroma, reflecting the basal vascular situation.
Downstream, the glomeruli are “ischemic”, with a retracted flocculus, enlarging the urinary chamber. Over time, the cellularity of the flocculus decreases, fibrosis lesions appear, which gradually extend to all the flocculus; this glomerulus then evolves towards the aspect of “bread to be sealed”.
In parallel with the formation of this glomerulosclerosis, tubular atrophy and fibrosis of the interstitium are generally observed. This fibrosis is often the site of lymphoplasmocytic infiltrates.
These lesions are very similar to those observed during renal aging, even in non-hypertensive subjects.
Hypertension could simply amplify this physiological phenomenon and advance it over time. But the diagnosis can also be disputed with certain segmental and focal hyalinoses. The more advanced the lesions, the more the diagnostic certainty is fragile.
Risk factors:
In populations, the renal risk is related to the level of arterial pressure:
Various studies have considered in a population subjects becoming renal failure, and analyzed the parameters separating them from the rest of the population. Blood pressure occupies a prominent place, which is not the formal proof of filiation.
According to the NHANES study, 3% of the American population has a high creatinine (at the threshold of 141 μmol for men and 124 for women). Among these subjects, 70% are hypertensive and 75% of the hypertensives are treated.Higher pressures are associated with a higher frequency of renal failure. The current treatment is generally insufficient, the blood pressure (PA) under treatment remaining well above the values recommended by the recommendations.
Since it is commonplace when the control of PA is defective, 48% of these subjects are in monotherapy.
Tozawa et al. followed 98,000 subjects from the general population of Okinawa, Japan, who had had a medical evaluation in 1983. After 17 years, 0.4% of these subjects were dialysed. The risk of such an evolution was, again, closely correlated with PA at the beginning of the study. It should nevertheless be mentioned that 5% of these subjects were already proteinuric at first, and even 16% if they were carriers of so-called severe hypertension, which leaves a serious doubt about the filiation of the events. For this reason, a statistical adjustment for proteinuria was made, and the relationship with PA persisted despite this adjustment.
Other studies of unselected populations have similarly found a relationship between the level of blood pressure and the risk of renal failure. In the Washington County study (23,500 subjects), the relative risk of having nephropathy increased with hypertension.
Compared to subjects with “optimal” blood pressure (<120/80) this risk was 3.8 (0.8-17.2) for grade 1 hypertension, 6.3 (1.3-29) for grade 2 hypertension, and 8.8 (1.8-43) for grades 3 and 4. In the Framingham cohort, 20 years of renal failure (defined as estimated creatinine <60 ml min -1 ) was assessed for all parameters of cardiovascular risk.
Hypertension had an odds ratio of 1.57 (1.17-2.12).
The meaning of these data is however limited. It merely states that in any population, patients with renal insufficiency are likely to be hypertensive and generally poorly controlled. What’s more, hypertension integrates with other factors.Age and possible diabetes have a greater statistical weight. The level of creatinine (or its clearance) at the beginning of the study is also a major determinant in all studies. For example, in the Framingham cohort, the odds ratio was 2.36 for age, 3.01 for an initial clearance of less than 90 ml min -1 , 2.6 for diabetes, and the 1.57 associated with hypertension did not fit into the multivariate model.
Risk factors in hypertensives:
Fewer studies were based on the hypertension phenotype, and these patients were at higher risk of developing renal failure.
That of Perry et al. is, in this respect, exemplary. These authors studied a cohort of 12,000 US veterans, 52 years old on average at the start of the study, and followed for at least 14 years. All subjects had uncomplicated hypertension when included in the study.
Significant risk factors for onset of renal failure were black race, diabetes, history of stroke (stroke) or (poorly defined) urologic “problem”. Age and smoking did not have a significant impact.
Blood pressure figures before treatment had a major impact. The incidence
end stage renal disease (CKD) is ten times greater if the initial systolic blood pressure (SBP) is more than 180 mmHg, compared to subjects for whom it is less than 165 mmHg. Values of the same order were found for diastolic, mean pressure, or pulsed pressure. The level of each of these parameters under treatment also had a significant impact on the risk of renal failure. The relative risk of renal failure was 3 (2.09-4.55) for patients whose PAS remained above 150 mmHg compared to those for whom it was less than 125. Having a A decrease of more than 20 mmHg in treatment-related PAS was associated with a relative risk of 0.39, that is, divided by almost three the risk of renal failure. Finally, the occurrence of myocardial infarction or heart failure during the study period greatly increased the risk of renal failure.
Vupputuri et al. followed a cohort of 722 hypertensives (including 504 blacks). They found that each standard deviation upwards of PAS (18 mmHg) and diastolic blood pressure (DBP) (11 mmHg) was associated with an annual decline in glomerular filtration of -0.92 and -0.83 ml min -1 respectively.
Patients whose blood pressure was above 160/95 had a relative risk of 5.21 for early renal function decline.
Similar findings have been made on populations included in large therapeutic studies. In the population examined for inclusion in the MRFIT study, after all adjustments for age, ethnicity, socio-occupational category, cholesterol, diabetes, the relative risk of end-stage renal failure was 5 (3.7- 6.7) for the top quintile of PAS. The renal survival curves as a function of the blood pressure level were similar to those of Perry. Similarly in SHEP, among 2,181 subjects over 65 years of age with pure systolic hypertension (placebo arm of the study), the relative risk of renal failure was 2.44 for the highest pressure quartile arterial compared to the lowest.
As already mentioned, European studies have provided different elements. Thus, in a British cohort of 176 patients followed for 12 to 14 years, there was no renal failure, let alone terminal, regardless of the severity of hypertension. In a Swedish cohort of 686 patients followed for 20 years, only 1.7% of patients had a modest elevation of creatinine (139 μmol l -1 ) without further explanation, and no end-stage renal failure was observed. Even in New York where Madhavan et al. followed for 5 years 2,125 patients with mild to moderate hypertension. Less than 2% of these patients achieved a plasma creatinine of 177 μmol or higher. Although the follow-up was relatively short, the authors insist that this small group of patients who developed renal insufficiency (3% of black subjects and 1.4% of white subjects) was distinguished by the existence of a initial proteinuria in 31% of cases. The authors concluded that subjects who increased creatinine within this time frame were likely to have pre-existing nephropathy.
Pathophysiology: renal consequences of arterial hypertension
As in many other fields, it is the data obtained in animal experiments that provide some certainty about the mechanisms by which hypertension can cause kidney failure. It is then possible to draw some conclusions about the findings made in human pathology.
The renal consequences of hypertension are related to three factors:
• the level of systemic blood pressure;
• the degree to which it is transmitted to the glomerular circulation;
• tissue factors of sensitivity to “barotrauma”.
Animal experimentation:
Mechanical effect of pressure:
The relationship between blood pressure and renal vascular injury has been extensively studied in experimental models.
The most classic in this respect is Goldblatt’s “two kidneys one clip” model, thus having a stenosis of one of the two renal arteries. The advantage of this model is that the kidney downstream of the clip does not undergo the effect of the increased pressure, only the intact contralateral kidney being exposed. On this side, hyaline, segmental lesions appear very rapidly, expressing plasma exudation and focal necrosis of the media.
Interestingly, these lesions appear even before hypertension is established (but continuous measurements show instability of blood pressure at this very early stage, with many blood pressure spikes). About 2 weeks after the clip is applied, the wall-light ratio of the vessels is increased by thickening the media. The necrotic lesions are then more rare in the interlobular arteries, suggesting that the hypertrophy of the media has a certain protective effect. Over time, the wall thickening continues and glomerular ischemia and sclerosis appear. Another highlight and relevant to the human clinic, the removal of the clip after a few months no longer stops hypertension. It is therefore self-maintained by the vascular lesions of the contralateral kidney. And in fact, if we remove it, the PA returns to normal. These facts illustrate the phenomenon of autoaggravation of hypertension and renal vascular lesions.
Self-regulation:
This mechanical effect of pressure on glomerular vessels is in fact singularly nuanced by the degree to which changes in systemic pressure are transmitted to the glomerular circulation. Indeed, these are normally amortized by a myogenic response of the afferent arteriole, under the dependence of NO. This response is potentiated by tubuloglomerular feedback from the macula densa. Any increase in blood pressure is accompanied by a vasoconstriction of the afferent artery that dampens its transmission downstream. This results in self-regulation of the glomerular pressure and flux, the protective effect of which is obvious.
This self-regulation is itself largely modulated by genetic factors. It is for example defective in a strain of spontaneously hypertensive rats, so-called fawn-hooded.
These animals, whose resistance to the afferent arteriole is very low, develop a severe nephroangiosclerosis early despite very modest blood pressure figures. Van Dokkum et al. have shown that there are at least two genes that condition the susceptibility of these animals to kidney failure. Their loci have been identified and differ from those controlling hypertension. The F1 hybrids of fawn-hooded rats with normotensive animals of ACI strain (thus heterozygous for these two genes) are, at identical level of blood pressure, protected from glomerulosclerosis resulting from hypertension induced by a non-selective NO inhibitor. -synthase, L-NAME.
In contrast, SHR strain rats, which have afferent arterioles of reduced light, are very insensitive to the renal effect of hypertension and develop little glomerulosclerosis.
Churchill et al. transplanted a kidney from a Brown-Norway (BN) normotensive rat to replace one of the two kidneys of a hypertensive animal (SHR), and induced hypertension in the recipient by administration of salt and DOCA. This hypertension induced proteinuria, decreased glomerular filtration, and glomerulosclerosis in the transplanted BN kidney and not in the native SHR kidney.
Self-regulation can be modified by pharmacological agents. Thus, in SHR, ACE inhibitors retain self-regulation, and even tend to lower glomerular pressure by dilatation of the efferent arteriole. In contrast, calcium channel blockers abolish self-regulation. The glomerular pressure then becomes strictly dependent on the systemic pressure.Glomerulosclerosis lesions induced by hypertension are then strongly increased.
Finally, self-regulation is highly compromised when the kidney is hyperfiltered. The best experimental model is 5/6 nephrectomy, which causes major hyperfiltration of the remaining nephrons, as well as increased nephron blood flow.In this model, the constitution of glomerulosclerosis is rapid and the sensitivity to variations of extreme pressure. The beneficial effect of suppression of the renin-angiotensin system, which normalizes glomerular pressure, is particularly dramatic. The absorption of a low-protein diet preserves self-regulation and limits glomerulosclerosis lesions despite the persistence of hypertension. Again, the addition of a calcium blocker cancels this effect and the protection induced by dietetics is abolished.
Tissue susceptibility factors:
It is impossible to explain the totality of the experimental (and human) findings without admitting that other factors, independent of the pressure, induce a particular susceptibility of the glomeruli. These factors are not clearly identified.They act by a common pathway of induction of free radicals generating oxidative stress, activation of growth factors, and mediators of fibrogenesis, such as transforming growth factor- b (TGF- b ) and plasminogen activator inhibitor 1 (PAI-1). A deleterious action on the podocytes, altering the mechanical support that these offer to the glomerular structure, was also evoked.
The induction of these factors has been largely attributed to the renin-angiotensin system. In fact, in addition to its own pressurized and hemodynamic actions, angiotensin II activates renal fibrogenesis, stimulates the production of TGF- b, increases oxidative stress, and finally stimulates various local mesangial inflammation factors. It is not easy to separate, within the many actions of angiotensin II, what is or is not dependent on pressure. Nevertheless, recent work suggests that some of these actions are effectively independent of the pressure.
Aldosterone is the latest of the offending factors. It has long been known that in the subtotal nephrectomy model, treatment with an angiotensin converting enzyme inhibitor can significantly reduce proteinuria and the development of glomerulosclerosis lesions.
In this situation, an aldosterone infusion restores the progression of these lesions despite the persistent blockage of the renin-angiotensin system. Conversely, blockade of aldosterone receptors has a good preventive effect on this type of nephropathy. Aldosterone could thus be a factor in renal fibrogenesis.
Rocha et al. have shown convincingly that spironolactone in the hypertensive rat “stroke prone” (SHRSP) significantly reduces proteinuria and the progression of glomerular fibrous lesions by an action independent of arterial pressure.
Aldosterone appears to also exert “non-genomic” actions. It induces vasoconstriction of the afferent arteries, not blocked by spironolactone, possibly inhibited by nifedipine, and reproduced by inhibition of NO synthesis.
Human data:
Stage “functional”:
Renal blood flow:
The renal blood flow is usually frankly lowered in severe hypertension. The findings are much more variable in “mild” arterial hypertension (hypertension). It can be normal, lowered or even sometimes increased. It is at the earliest stage, or even before hypertension in susceptible subjects, that an increase has most often been observed.
This is almost never the case when hypertension has been around for some time. In a classical work, G.
London et al. measured renal blood flow in 131 patients with moderate uncomplicated hypertension and 67 normotensive controls. Flow was decreased in hypertensive patients compared to normotensive patients (739 versus 891 ml min -1 m -2 ). Moreover, the renal fraction of the cardiac output was also lowered, while the hepatic or muscular fractions of the cardiac output remained unchanged. There is therefore a specifically renal ischemia in the hypertensive. In these subjects, the alteration of the renal blood flow is all the more important as the figures of arterial pressure are high, it also increases with the age of the subject and increases with the time of evolution. It should be noted that it is reversible under treatment, particularly by an IEC, which indicates its functional character, at least at this stage.
Glomerular filtration:
Glomerular filtration is paradoxically long preserved in these subjects. In the presence of a lowered blood flow, the maintenance of the glomerular filtration supposes an increase of the filtration fraction, thus an increase of the intraglomerular pressure. This increase is minimal at the beginning and increases with decreasing renal blood flow.This compensation explains why the obvious anomalies of renal hemodynamics are not apparent for a long time on the simple biological assessments performed in these patients.
How is renal resistance distributed? Decreased blood flow despite high pressure indicates that renal resistance is generally increased. The fact that the filtration does not follow the pressure indicates that the resistance is largely preglomerular. Kimura et al. have verified this increase in afferent arteriolar resistance, with glomerular pressure and efferent arteriolar resistance still normal in essential hypertensives. On the other hand, the increase in the filtered fraction found by London suggests that there is also a modulation of the postglobular resistance.
More recently, Safar et al. showed that there is a correlation between creatinine clearance and arterial stiffness as measured by pulse wave velocity in subjects with normal or subnormal renal function. This relationship is independent of mean arterial pressure (MAP), and other cardiovascular risk factors. It indicates that the link between renal pathology and hypertension is not limited to arterioles of resistance, but also involves large elastic arteries.
The appearance of microalbuminuria is often the first biological marker of renal impairment in hypertensives. The mechanism of its occurrence is still debated. Is this a simple “mechanical” consequence of the increase in intraglomerular pressure, is it endothelial dysfunction (probably reflecting a much more diffuse mechanism), or a first marker of an anatomical lesion? Its reversibility when lowering the AP would be more in favor of the first hypothesis.
Self-regulation:
Some findings in human pathology are very close to the knowledge acquired in animal experiments. Thus, for identical pressure, the renal consequences of hypertension appear much greater if there is a pre-glomerular vasodilatation, such as is observed for example after a nephrectomy or in type I diabetes. Still in these cases is moderate vasodilatation, with relatively preserved self-regulation.
This is probably the reason for the modest consequences of unilateral nephrectomy and the long delay in onset of diabetic nephropathies. The lesions appear and increase much more rapidly when the nephron reduction is greater.
Still, it is probably necessary to distinguish the continuous or average level of PA and the instability of the pressure with peaks; each being capable of inducing kidney damage.
There is also obvious genetic influence in humans, as evidenced by the familial aggregation of cases of end-stage renal failure in subjects with nephroangiosclerosis. It is also known as much more common in black subjects than white. However, it has been shown in black subjects a lesser capacity for self-regulation.
Kotchen et al. studied black, Caucasian, hypertensive subjects who were subjected to increasing doses of norepinephrine. Black subjects had higher baseline renal blood flow and glomerular filtration than white, indicating relative hyperfiltration.
Under the influence of norepinephrine, the renal plasma flow remained unchanged in both groups. In contrast, glomerular filtration was strongly increased in black subjects while it remained stable in white subjects.
Fernandez-Llama et al. studied the distribution of insertion-deletion (ID) polymorphism of the angiotensin-converting enzyme gene in 37 subjects with histologically proven nephroangiosclerosis. They showed a strong asymmetry of this distribution with a more frequent DD genotype (57% compared to 25% in the control population) and a rarer DI genotype (27% versus 64%). There was no difference in distribution between hypertensive patients with normal renal function and normotensive controls. These authors concluded that this polymorphism could be a marker of renal susceptibility to hypertension.
Recently, Fabris et al. studied the four main polymorphisms known to affect the renineangiotensin-aldosterone system (angiotensinogen, converting enzyme, AT1 receptor and aldosterone synthase). They found a relationship between each of these polymorphisms and the existence of renal failure in hypertensive subjects.
They also found interactions, positive or negative, between the different polymorphisms on this same criterion.
Many other candidate genes have been mentioned, including endothelin, NO-synthase, TGF- b , platelet derivated growth factor (PDGF) … their study has not yet made it possible to decisive conclusions about a dominant role of one of them.
Lesional stage:
These renal functional abnormalities leave place, with the time, with anatomical lesions and this, all the more easily as the glomerular circulation is more exposed and that the glomerular pressure is higher. The direct responsibility of the mechanical pressure factor in the genesis of vascular lesions on the one hand, and glomerular lesions on the other hand, can be intuitively accepted by analogy with what is observed in animals, particularly in the Goldblatt model. It is nonetheless difficult to prove, and its exclusive role seems very unlikely. On the one hand, human hypertension is a complex situation that involves a whole series of abnormalities that can, by themselves, generate vascular lesions or even glomerular lesions. On the other hand, the obvious lack of correlation between the level of pressure and the existence of these lesions indicates quite well that the intervention of other factors is necessary for the appearance of nephroangiosclerosis.
Nephroangiosclerosis, a diagnosis error?
In the past 10 or 20 years, data have accumulated showing that the field of renal injury “induced” by hypertension extends far beyond “nephroangiosclerosis” in its classical sense. The existence of hypertension plays a considerable role in aggravating any nephropathy, whatever its origin. However, many nephropathies, even at a very early stage, and almost all at an advanced stage, are accompanied by hypertension. This, in turn, precipitates the evolution of kidney damage.
If we take into account the absence of relatively specific clinical symptoms, a usually inaccurate chronology and the fact that a renal biopsy is very rarely performed in these patients (again the histological lesions are not specific), it is easy to understand that the mere filiation of hypertension generating kidney damage may seem a bit simplistic. The alternative hypothesis is that renal lesions pre-exist, are responsible for hypertension, and that there is a vicious circle of autoaggravation.
Fogo et al. have thoroughly analyzed the renal histology data in patients in the AASK study. This study involved African American hypertensive subjects who were considered to have nephroangiosclerosis. Of the 39 biopsies, 38 showed a significant degree of arteriology or arteriolosclerosis, authenticating the diagnosis. Other lesions, however, coexisted either with segmental hyalinosis of the glomeruli or clearly of other nature, including cholesterol emboli.Later work in the same group compared these lesions with those seen in Caucasian hypertensives, and showed significant histological differences. In particular, black subjects were more likely to have a “solidified” form of glomerulosclerosis, with white subjects mostly having an “obsolescent” form. These lesions were correlated neither with age nor with PA, and very little with proteinuria. The authors concluded that these different phenotypes probably corresponded to different lesional mechanisms and that their data “do not support the idea of a direct causal link, as suggested by the term nephroangiosclerosis, according to which hypertension is responsible for of vascular sclerosis which, in turn, causes glomerulosclerosis “.
Conversely, various histological studies have been reported previously in non-renal and normotensive patients. These were either isolated proteinuria or young women with pre-eclampsia who wanted to eliminate another kidney disease.In both cases, normotension was firmly established, renal function was normal, and the biopsy gesture was intended to be prognostic. In a large number of cases, only vascular lesions have been observed, with or without hyalinization of the vascular poles of the glomeruli. This simple observation indicates that such lesions are compatible with strict normotension. Moreover, in the follow-up of young women after preeclampsia, the patients carrying these lesions have most often become hypertensive after a few years, which is very atypical, especially in young women.
These facts strongly suggest that nephroangiosclerosis lesions preceded hypertension.
Over time and successive arguments, doubt has evolved into a strong skepticism, leading some authors to believe that the idea that a non-malignant hypertension can itself be responsible for end-stage renal failure is not based on anything consisting. Critical analyzes of the concept of nephroangiosclerosis have since multiplied.
Approximate diagnosis:
As mentioned above, the sequence of events is usually imprecise, not only in clinical practice but even in the studies that are supposed to refer to it. There are, in fact, few studies in which the absence of proteinuria and the normality of creatinine clearance were established during the discovery of hypertension. In clinical practice, renal biopsy is – rightly – only rarely performed. As a result, many of the immunoglobulin A (IgA) nephropathies, segmental and focal hyalinoses, or even chronic interstitial nephropathies of any kind, are initially unknown and then considered as nephroangiosclerosis, as they have not been properly labeled in a timely manner. All the statistical data which we have mentioned mention a nephropathy of hypertensive origin solely because no other cause than hypertension is known.There is therefore every reason to believe that there is a significant overestimation of this diagnosis. And indeed, all the studies that have looked into this problem confirm the frequency of clinical misdiagnoses.
Schlessinger studied 43 kidney transplant patients who had been “unambiguously” diagnosed with nephroangiosclerosis. Of the few patients who had had a renal biopsy, none had the histological features of benign nephroangiosclerosis. In addition, it was possible to document the coexistence of hypertension and renal function that is still normal in less than 5% of cases. For Kincaid-Smith, the study of kidneys removed during transplantation in equally-diagnosed patients has only exceptionally confirmed it.
Caetano et al. have reported the histological renal study of 81 hypertensive patients with moderate renal impairment.Benign nephroangiosclerosis was retained as a histological diagnosis in 22% of cases. Atypical vascular lesions, suggesting episodes of malignant hypertension, were considered in 43% of cases. Other nephropathies, primary glomerular, were involved in 35% of cases (about half of segmental and focal hyalinoses).
A Swedish study, already mentioned above, prospectively followed a cohort of 686 hypertensive patients over a period of 20 years, with careful monitoring of renal function. An increase in creatinine was observed in 8.9% of these patients, but almost all of them were found to have other types of nephropathy. These included various primary nephropathies, diabetic nephropathies, urological obstructions or renal arterial stenoses. Only 12 patients (1.7%) had a very modest elevated creatinine (139 μmol l -1 at 20 years of follow-up) with no explanation other than hypertension.
Zarif et al. studied the population of nine dialysis centers in Ohio. Of these, 220 (37%) were known to have nephroangiosclerosis as their nephropathy. Four of these had been biopsied, one had compatible histology, the others had IgA nephropathy, segmental and focal hyalinosis, and chronic interstitial nephropathy. The authors tested in others the criteria of “phenotyping” of hypertensive nephropathy.
In more than 50% of cases, the available data were insufficient to make a decision, and for 14% it could only be a misdiagnosis. Erroneous diagnoses spanned all nephrology, glomerular or interstitial, to a kidney of myeloma and nephropathy of the human immunodeficiency virus (HIV).
Ischemic nephropathy:
The qualified entity of ischemic nephropathy is easily confused with nephroangiosclerosis and constitutes an important diagnostic alternative. In principle, however, it is very different. Ischemic nephropathy consists of lesions downstream of more or less well-characterized stenoses of the renal arteries, resulting in defective perfusion of the parenchyma, possibly below the threshold of self-regulation. This ischemia activates the renineangiotensin system, which contributes to the increase in BP and tends to establish a vicious circle with vascular lesions. Its mechanism is therefore very different from that of nephroangiosclerosis.
The use of antihypertensives must be more nuanced because the risk of aggravating ischemia is obvious. The alteration of renal function justifies the discussion of an angioplasty, a debate in which we will not enter here. Finally, ischemic nephropathy is totally in the context of diffuse atheromatous disease and its treatment is largely that of vascular risk. Moreover, if the stenosis (s) have not been diagnosed, the patient suffers from renal insufficiency, a priori of vascular origin, whereas he has been hypertensive for a long time. And it is clear that at an advanced stage of renal failure, the distinction between these entities becomes more and more difficult, including in the event of renal histology.Thus Zucchelli et al. reviewed the renal biopsies of 56 consecutive patients who had been diagnosed with nephroangiosclerosis. This diagnosis was consistent in only 48% of cases, while 35% had atheromatous renal vasculopathy and / or cholesterol embolism.
Nephropathy responsible and not consequence of arterial hypertension?
The idea is defended with more and more insistence – and arguments – that the primary anomaly causing hypertension could be very often renal (the “guilty kidney”).
The mechanisms proposed are diverse and not exclusive, and it is likely that each of them can logically involve only a subgroup of patients. Nevertheless, there are important hyphens between them: these abnormalities are for the most part progressive and may therefore explain the progressive worsening of hypertensive nephropathy.
This evolution has the common denominator hyperfiltration, ultimately major source of glomerular sclerosis. The kidney is much more vulnerable to pressure variations when it is hyperfiltering, it is easy to understand the ensuing autoaggravation loop.
A major reason for considering kidney as the origin of hypertension lies in transplantation experiences.
Much work has been done since Dahl’s initial publication, which showed that “hypertension follows the kidney”. In many models of genetic hypertension, if a kidney from a hypertensive donor is transplanted to a normotensive recipient, the latter becomes hypertensive, and vice versa. In human kidney transplantation, such sharp data are impossible to obtain, but it is clear that hypertension appears more often in kidney recipients of a hypertensive donor than if the donor was normotensive.
Genetic anomaly of sodium reabsorption:
Essential hypertension is readily associated with a disorder of renal excretion of sodium (Na). Everything suggests that this disorder is constitutional, and concerns the proximal reabsorption of sodium. Among the various hypotheses of genetic anomalies that may explain this disorder, that of a mutation of one or more of the three subunits of α- aducine is undoubtedly the most consistent and the most advanced. Very early, elegant theories have suggested that hypertension may be the haemodynamic price to pay in the presence of such a disorder for the maintenance of a balanced sodic balance.
Glomerular hyperfiltration may be the first term of this compensation. Indeed, if there is an excess of proximal reabsorption, limiting sodium intake to the distal nephron, the introduction of tubuloglomerular feedback gives rise to a compensatory increase in glomerular filtration. In fact, Bianchi has shown that glomerular filtration is higher in normotensive young subjects whose two parents are hypertensive (thus a priori predisposed to hypertension) than in those with both parents being normotensive.
This idea has been reinforced by more recent work. Barba et al. studied glomerular filtration and proximal sodium reabsorption in 47 healthy normotensive volunteers, which they classified according to the sensitivity of their AP to changes in sodium intake. Sodium-sensitive subjects had the highest AP, although this remained within the normal range.
In the normosized diet, these subjects had both higher glomerular filtration and higher proximal sodium reabsorption than the others. The difference in glomerular filtration disappeared in a diet low in salt. This same finding of increased pressure and glomerular filtration in sodium-sensitive subjects was made by Weir.
Kotchen’s work, already mentioned above, showed hyperfiltration in hypertensive black American subjects and the same finding was made by Parmer. Palatini et al. recently confirmed hyperfiltration in young subjects with limited hypertension, especially if they had microalbuminuria.
The idea is thus widely accepted that hyperfiltration is common at the earliest stage of a hypertensive disease and that it is even likely to precede the increase in BP. As we saw above, this hyperfiltration only has a time, and gradually fades when hypertension has been installed for years.
A number of nephrons reduced at birth?
Brenner suggested that hypertension could be linked to a reduced number of nephrons at birth, responsible for compensating hyperfiltration. This would then explain not only the onset of hypertension, but also the subsequent nephropathy.
Very strong arguments have been developed in favor of this idea. Among various studies, particularly convincing, is that recent, in which were compared the kidneys of ten hypertensive men and ten others, normotendus, died in road accidents. The hypertensives had half as many nephrons as normotendus (702,379 versus 1,429,200), and their glomeruli were hypertrophic.
Moreover, counting and measuring glomeruli are not so simple a job. Hoy et al. reported a study of autopsy kidneys from a wide variety of ethnic groups. They observed a variation in the number of glomeruli by a factor of 9, and a variation in their volume by a factor of 5.6. There was no clear interethnic difference.
Nevertheless, the authors observed a strong correlation between the number and the volume of the glomeruli. This observation suggested that increasing the volume of glomeruli could be a compensating factor in reducing their number, and thus represent a susceptibility factor for renal failure.
Is a reduced number of nephrons sufficient to explain hypertension? Data obtained after unilateral nephrectomy in adults (kidney donors for example) are a little discordant, but overall, the prevailing impression is that this nephrectomy does not induce hypertension, even with a very long decline. Nephrectomy in very young children (most often because of a tumor) seems to be more likely to be accompanied by hypertension in adulthood. In the extreme case, in the rat, nephrectomy performed in the hours following birth leads regularly and rapidly to hypertension, whereas it has no effect later. These data suggest that not only is the reduction in the number of nephrons involved, but also the timing, and therefore the potential for compensatory hypertrophy of these nephrons, it is this that is the key hyperfiltration.
Programming during intrauterine life?
Barker has published a long series of works indicating that chronic fetal distress with hypotrophic birth is associated with a much higher incidence of hypertension and diabetes in adulthood. This concept has been validated by numerous studies, although it remains somewhat controversial.
Law and Shiell reviewed 21 studies that almost all showed lower PAS in adulthood for each additional birth weight bracket. This idea of ”perinatal programming” has given rise to many pathogenic hypotheses. This relationship has been attributed to fetal “malnutrition” related to either maternal malnutrition or poor placental hemodynamics. An alternative hypothesis would rather incriminate rapid growth (“catch-up” growth) following a hypotrophic birth. This accelerated growth is said to be a source of hyperinsulinism, and thus the bed of the metabolic syndrome and hypertension.
Experimentally, placental ischemia at the end of gestation (aortic clip) gives birth to children of reduced weight, who quickly develop hypertension. Similarly, several studies of maternal protein restriction in animals have shown that children born of these gestations had a reduced number of glomeruli, especially if the protein restriction focused on the end of gestation. It has also been shown that these smaller and larger glomerulus animals have early hypertension, proteinuria, an increased number of AT1 receptors for angiotensin.
Rodriguez et al. have shown in the human species a reduction in the number of nephrons in very premature babies.Similarly, Hughson et al. found a linear relationship between the number of glomeruli and the birth weight of the subjects. Hofman et al. showed the constant presence of insulin resistance in children aged 4 to 10 years born premature and / or hypotrophic.
We will not detail here all this argument, of which a general review was recently published. The general idea that emerges is that some individuals may be born with a reduced number of nephrons and compensatory hypertrophy.
This hyperfiltration would carry in itself both the germ of hypertension and that of glomerulosclerosis. The mechanisms of this programming appear complex and are not completely understood. They could involve the fetal renin-angiotensin system, glucocorticoid metabolism, and other systems during the corresponding period of nephrogenesis in humans in the third trimester of pregnancy.
Obesity, hyperinsulinism, hyperfiltration:
The description of a nephropathy related to obesity is relatively recent. This nephropathy is essentially a segmental and focal hyalinosis, with the particularity of large glomeruli. The frequency of this nephropathy is of the order of 2% of kidney biopsies, but it has increased tenfold in just over 10 years, raising the possibility of an “epidemic” situation in the United States. The privileged coexistence of hypertension with obesity, or even the complete metabolic syndrome, has raised the hypothesis that this nephropathy may be an integral part of the fuzzy set of “nephroangiosclerosis”.
The trait common to all components of the metabolic syndrome is insulin resistance, and this is an important factor in the development of nephropathy. Chen et al. showed in a population of 6,400 non-diabetic subjects from the NHANES 3 study, a close link between the existence of nephropathy (creatinine clearance <60 ml min -1 ) and the plasma level of insulin, peptide C, hemoglobin A1c and the insulin resistance test. The same authors showed a relative risk of nephropathy of nearly 3 in subjects with a metabolic syndrome, and a correlation between this risk and the number of components of the metabolic syndrome present. Chagnac et al. have shown that obese subjects have a renal blood flow increased by vasodilation mainly on the afferent arteriole, and an increase in glomerular pressure. A parallel increase in glomerular filtration is associated with it.
It is correlated with the degree of insulin resistance. These facts seem to indicate, once more, the central role of hyperfiltration and afferent vasodilation in the genesis of glomerular lesions.
This idea is not only of theoretical interest. Indeed, an early and intensive intervention on overweight should be able to significantly limit the evolution of this nephropathy. In addition, she could discuss the value of treatment with peroxisome proliferator activated receptor (PPAR) agonists, known to have a favorable effect in the metabolic syndrome.
Acquired microvascular disease of the kidney: the related arteriole
Hypertensive subjects very often have arteriolar, renal and systemic lesions, relatively diffuse. We have seen that such lesions may well precede the elevation of AP. These lesions have been widely described in the past as arteriolosclerosis. These findings have led Goldblatt to postulate that such lesions may be primitive, and secondarily responsible for hypertension by an ischemic mechanism since they largely affect the related arterioles. The possibility of experimentally reproducing such lesions, and the observation that they are followed by the appearance of salt-sensitive hypertension, has given new relevance to this theory. The mechanism by which these lesions are created, however, remains rather mysterious. Inflammatory phenomena, mononuclear cell infiltration and transient spikes of catecholamines have been reported, generating brief, jolting jerks. In recent years, uric acid has also been implicated through a direct effect on the proliferation of smooth muscle cells and endothelial dysfunction.
Synthesis:
These data are relatively disparate and refer to multiple hypotheses. We will certainly not try to draw up a unicist schema. And indeed, not all hypertensives are dependent on salt, not all hypertensives are born hypotrophic or malnourished mothers, etc.
It can nevertheless be seen that a whole bundle of arguments converges towards an abnormality of renal physiology, acquired before birth. This anomaly can be either constitutional and genetic, or acquired during intrauterine life because of an unfavorable maternal environment, it includes in this case an oligonéphronie. In both cases, the situation is characterized by a difficulty of equilibration of the sodic balance. This is compensated by glomerular hyperfiltration, either absolute in the case of a genetic anomaly, or with a hypertrophy of the remaining nephrons.
This hyperfiltration is in turn generating, according to Brenner’s model, hypertension, proteinuria and glomerular hyalinosis lesions.
A role of hyperinsulinism and the metabolic syndrome also has strong arguments in support. When is this situation “programmed”? From the intrauterine life for some, during the “catch-up growth” of the first years of life for others, perhaps later, under purely or essentially environmental influences.
But in any case, in this hypothesis, the individual will be, in adulthood, hypertensive, obese, carrying a metabolic syndrome, and a nephropathy that will be called nephroangiosclerosis since it There is no other obvious cause than hypertension.
As for the primary lesions of the afferent arterioles, they could be integrated in the same pattern or, opposing the “Brennerian” dogma, have totally different mechanisms, immunological, inflammatory or other. This theory is certainly seductive, but expects to have more consistent arguments.
Treatment:
Any therapy should satisfy several imperatives: lowering the pressure load certainly, but also normalize the renal hemodynamics and in particular the glomerular pressure, respecting the self-regulation of the afferent artery. All experimental and clinical data indicate that the patient is at increased risk of progression to renal failure because proteinuria is important (probably reflecting the degree of renal hemodynamic disturbance), and correlatively the beneficial effect. treatment is largely dependent on the reduction of proteinuria that it causes. Therefore this criterion is considered absolutely essential in the choice of a drug in these subjects.
Low blood pressure:
The basis of treatment, both preventive and curative, is the standardization of PA. The emergence of effective antihypertensive treatments has almost eliminated malignant nephroangiosclerosis and appears to have significantly reduced benign nephroangiosclerosis. The strong relationship between AP level and the likelihood of an IRT suggests that maintaining normotension can largely prevent the deterioration of renal function. Several studies have considered the tension figures under treatment to establish this relationship. It is more rare that the reduction in AP has itself been taken into account. In Perry’s study, pressure reduction was clearly associated with a decreased risk of kidney failure.
Hsu criticized this equation with some virulence.
He conducted a meta-analysis of 10 prospective controlled PA reduction studies that lasted more than 1 year. The relative risk of developing renal insufficiency (patients treated versus controls) was 0.97 (0.78-1.21). The author concluded that antihypertensive treatment has no nephroprotective effect.
It must be admitted that the studies considered were not designed to test this hypothesis, and that the incidence of renal insufficiency was very low, between 1 per thousand and 2%. For example, in the Syst-Eur study, there were three and two renal insufficiencies for 2,398 and 2,297 patients. There was therefore relatively little chance of observing a significant effect of the treatment, and the statistical power could not be sufficient despite the large number of patients.Moreover, almost all patients in these studies had very moderate hypertension, and severe cases were most often excluded, which greatly limited renal risk. Studies based on patients with known renal impairment were excluded from this meta-analysis. Such studies have produced results in a very different sense.
The MDRD study compared the decline in glomerular filtration in 840 patients with various nephropathies (insulin diabetics excluded) for two levels of AP used as a therapeutic target (MAP of 107 and 92 mmHg respectively for subjects under 60 years of age). ). The nature of the prescribed antihypertensive treatment was not controlled. The decline was more rapid in patients with the highest PA treatment, the relationship being stronger if the baseline proteinuria was higher.
The consensus reported in 2000 by the National Kidney Foundation (NKF) reports a linear relationship between the level of BP achieved during treatment and the annual loss of glomerular filtration in nine controlled studies in both diabetic and non-diabetic subjects. The minimum loss is clearly at a level of 98 mmHg PAM (130/80 mmHg) or less. It is three times higher for a MAP of 106 mmHg (140/90). This work is one of the foundations of the current recommendation of low pressures in nephropathic subjects, diabetic or not.
An exception in this unanimity is the AASK study. This very rigorous study involved 1,094 African-American subjects with nephroangiosclerosis, which was histologically confirmed for a significant number of patients, as mentioned above. The study involved a comparison of two PA targets, resulting in one group that reached 128/78 mmHg and the other 141/85 mmHg. There was no difference between these groups on the glomerular filtration decline, nor on the composite endpoint with a loss of 50% or more glomerular filtration, end-stage renal failure or death. In addition, proteinuria was lower throughout the study in the lowest BP group.
All current recommendations indicate a target value of less than 140/90 mmHg in “allergic” hypertension, which is lower in cases of diabetes or nephropathy. It is known that this target is not reached in the majority of patients, so far, strong efforts of persuasion remain necessary both for patients and their doctors. If self-regulation of the efferent arteriole is compromised in subjects with nephropathies and in diabetics, the transmission of systemic pressure to the glomerular circulation is greater in these subjects.
This is why PA should be lowered at even lower values to avoid worsening of nephropathy.
Inhibition of the renin-angiotensin system:
The idea that an equal fall in PA, the inhibition of the renin-angiotensin system by a converting enzyme inhibitor or by an angiotensin II receptor antagonist has a specific nephroprotective power flowed a lot of ink for a decade.
The pathophysiological foundation is well established. Angiotensin II causes preferential vasoconstriction of the efferent arteriole. For this reason, it increases the intraglomerular pressure and the filtration fraction. This increase is a factor in safeguarding glomerular filtration during a decrease in renal plasma flow, but it is also, in the long term, the source of progressive glomerular sclerosis.
In the experimental model of nephron reduction, the administration of a conversion enzyme inhibitor dramatically reduces proteinuria and glomerular sclerosis, compared to any other antihypertensive treatment.
This favorable action is the corollary of a normalization of intraglomerular pressure.
In clinical practice, a specific nephroprotective effect of renin-angiotensin system inhibition was unambiguously shown in diabetic subjects, those with type I diabetes mellitus and nephropathy, and those with type II diabetes mellitus. nephropathy, or a simple microalbuminuria.
Inhibition of the renin-angiotensin system even prevents the occurrence of microalbuminuria. The concept of renal protection in diabetics therefore seems very firmly established and this level of evidence has justified explicit extensions of marketing authorization (MA) for various drugs that inhibit the renin-giotensin system in diabetic patients with at least one microalbuminuria.
In non-diabetic subjects, the AASK study showed a 36% reduction in glomerular filtration rate and 48% reduction in composite endpoint (loss of 50% or more glomerular filtration, IRT, or death) under ramipril in comparison with amlodipine or metoprolol, in subjects with proteinuria> 300 mg 24 h -1 . In the entire cohort (including normo- and microalbuminuria), the difference in glomerular filtration decline was not significant but that of the composite endpoint was. After adjusting for various covariates, the difference between ramipril and amlodipine was 36% for glomerular filtration decline and 38% for composite endpoint. This study therefore concluded that there is a significant beneficial effect of ACE compared to other classes of drugs in African-American subjects with nephroangiosclerosis.
The REIN study similarly showed that subjects with chronic nephropathy with proteinuria were better protected by a converting enzyme inhibitor against the onset of end-stage renal failure than they were with a so-called conventional.
A meta-analysis of 11 controlled trials in nephropathic and non-diabetic subjects showed a substantial benefit of angiotensin-converting enzyme inhibitors. Under this treatment, after adjustment for BP, the relative risk of IRT was 0.69 (0.51-0.94) and that of creatinine doubling 0.70 (0.55-0.90) . This effect was all the more marked as proteinuria was abundant and became very low for proteinuria less than 0.5 g 24 h -1 .
In order to obtain a more complete inhibition of the renin-angiotensin system, the combination of a conversion enzyme inhibitor with an angiotensin receptor antagonist has been recommended for the last ten years. Such an association makes it possible to obtain a better reduction of proteinuria, and probably a better preservation of renal function. The main clinical study in favor of such a hypothesis is COOPERATE. In this study, 336 patients were recruited, all with non-diabetic nephropathy, and 263 were retained after an 18-week run-in period. They received by lot either losartan, trandolapril, or the combination of both. The relative risk of achieving the primary endpoint (creatinine doubling or end-stage renal failure) in patients receiving the combination was 0.38 versus trandolapril alone or 0.40 compared with losartan alone.
Faced with this very complete argument, even idyllic, he is nevertheless a strongly discordant voice. Bidani and Griffin, in a series of studies, have tried to show that nephroprotection by inhibition of the renin-angiotensin system is in fact totally dependent on the level of PA reached, whether in clinical studies or in animal experiments. Their argument is based primarily on the MAPA data for clinical studies and the permanent records for experimental studies.
Nevertheless, they have remained quite isolated in this position, and their arguments can not be definitively convincing.
Other authors have suggested that drugs that inhibit the renin-angiotensin system would have a stronger effect on central blood pressure than on the humeral artery, unlike, for example, b- blockers. This difference could explain the nephroprotection related to these therapies by an effect not “beyond the arterial pressure” but simply “beyond the humeral artery”.
Antialdosterone:
Arguments, mostly experimental, indicate:
• that aldosterone probably plays an underestimated role in renal fibrogenesis;
• the use of ACE inhibits aldosterone only very transiently, an “escape” effect occurring after a few days or weeks.
Antialdosterone, spironolactone and eplerenone, showed a favorable effect on proteinuria and renal fibrogenesis, in SHRSP rats, and in the nephron reduction model.
The argument for a nephroprotective effect of antialdosterones in clinics is essentially based on this theoretical basis, as studies are rare. Chrysostomou et al. studied eight patients with enalapril proteinuria greater than 1 g 24 h -1 . In these patients, the addition of spironolactone lowered proteinuria from 3.81 ± 2.5 to 1.75 ± 1.02. Similarly, Sato et al.showed an additional lowering of proteinuria by the addition of spironolactone in 13 diabetic “aldosterone escape” patients to an IEC. In their recommendation, Wilmer et al. consider the use of antialdosterones as a level of evidence 2.
A reminder to caution is however necessary. Both ACE inhibitors, angiotensin receptor antagonists, and antialdosterones all increase serum potassium. Mixtures are not necessarily innocuous or risk-free, and serum potassium should be well monitored in these patients, whose already impaired renal function further increases the risk.Recall that the publication of the RALES study was followed by a sharp increase in hyperkalemia accidents.
Calcium inhibitors:
As mentioned above, calcium channel blockers appear to be less favorable to renal function because they more or less abolish autoregulation of the afferent arteriole tone and thus promote the transmission of systemic pressure to the glomerular circulation. . If the cause seems heard in animal experiments, the human data are more fragile.Theoretically, these drugs would be deleterious for renal function, at least if the control of PA is imperfect, because of this transmission of systemic pressure. This effect seems to be more pronounced for dihydropyridines than for other calcium blockers (the latter may even have an antiproteinuric effect according to some studies). Finally, the homogeneity of the dihydropyridine class in this respect is probably not absolute.
Various studies indicate that dihydropyridines reduce microalbuminuria less than an IEC. Ruggenenti et al., In an ancillary study of REIN, identified patients who received a calcium channel blocker. During the course of the study, these patients maintained superior proteinuria, unless they also received ACE inhibitors and / or if the treatment regimen was less than 100 mmHg, which validates the hypothesis quite well. theoretical. In the IDNT study, amlodipine did not reduce proteinuria or improve renal prognosis, unlike irbesartan. Similarly, in the AASK study, amlodipine did not reduce proteinuria, unlike ramipril.
Amlodipine induced a transient increase in glomerular filtration, but this disappeared over time, giving way to a better protective effect of ramipril.
In contrast, calcium channel blockers appear to have no negative effects when combined with a conversion enzyme inhibitor or angiotensin receptor antagonist.
For example, in the RENAAL study, the renal outcome was not different in patients receiving or not receiving a calcium channel blocker, the benefit of losartan being unaffected. It clearly indicates that:
• a dihydropyridine is superior to a placebo, because of the fall of PA (Syst-Eur);
• a dihydropyridine is less than an IEC (AASK);
• a combination of dihydropyridine and IEC is equivalent to IEC alone (Nephros), and superior to dihydropyridine alone (REIN).
Ultimately, if calcium channel blockers do not seem to share the beneficial effect of other classes (pressure-independent nephroprotective effect), there is no sound study indicating that they are likely to have a deleterious effect. in the medium or long term on renal function. Most authors therefore agree that they do not recommend the use of a calcium channel blocker as first-line therapy or monotherapy in patients with renal impairment. However, there is no restriction on the use of these drugs in combination with an antihypertensive inhibiting the renin-angiotensin system, or as part of a combination therapy, and it is known that it is very often needed to reach the PA target in these patients.
Conclusion:
Nephroangiosclerosis is typically the long-term consequence of hypertension and is reflected in chronic progressive renal failure. This finding is relatively common in black American subjects with severe hypertension, it is exceedingly rare in Caucasian subjects and / or with moderate hypertension. Both clinical and experimental evidence indicates that PA may actually have direct consequences on renal physiology and anatomy. In contrast, ethnic inequalities and poor correlation with AP figures indicate that the relationship is not as simple, and that many other factors are involved in the genesis of this nephropathy.
The concept itself of nephroangiosclerosis has been called into question, in favor of that of a congenital nephropathy of multifactorial origin, which would be a source of both hypertension and subsequent progressive renal lesions. The mechanisms of the genesis of this nephropathy during intrauterine life are the subject of intensive studies.
The reality is probably not so manichean and significant progress remains to be made to better understand the nature of this nephropathy associated with hypertension. Anyway, it is at least very clear that PA and kidney damage are engaged in an autoaggravation loop. In practice, treatment can – and must – break this vicious circle. This rupture begins with an optimal control of PA. In this regard, whether the renal lesion is initially a consequence of mechanical pressure or, on the contrary, that it has engendered it, does not fundamentally change the therapeutic attitude. Some antihypertensives, especially those that inhibit the renin-angiotensin system, appear to have additional nephroprotective power, beyond the pressure drop. Their rational use, most often in the context of a combination therapy, should further reduce the frequency of renal failure in hypertensive patients, especially in exposed populations.