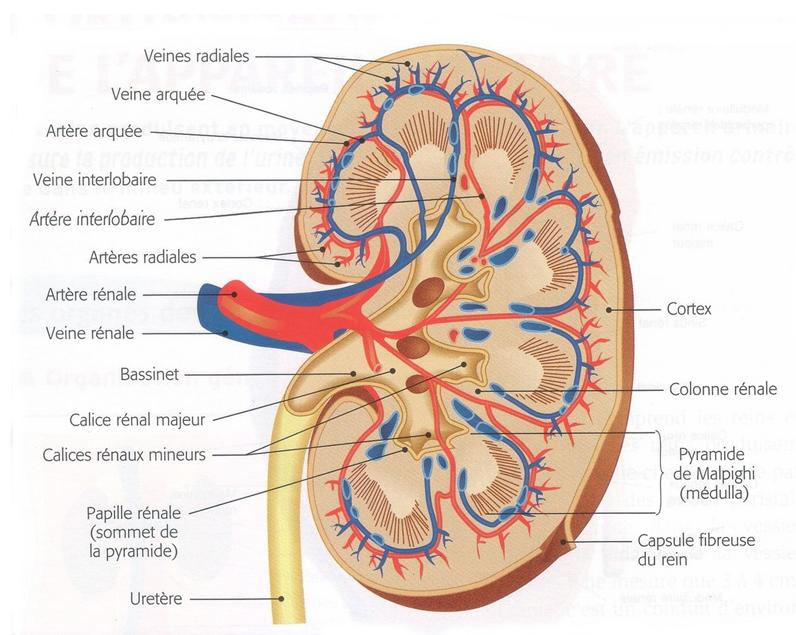
 GLOMÉ RULES AND GLOBAL FILTRATION:
GLOMÉ RULES AND GLOBAL FILTRATION:
Constitution of the primitive urine:
Renal blood flow accounts for 20% of the cardiac output and crosses almost all the glomeruli. The first step in the development of urine is the formation of the glomerular ultrafiltrate by diffusion of the plasma components through the glomerular filtration barrier. This diffusion is done as a function of the permeability and the selectivity of this barrier and according to the pressure gradients exerted on both sides. The fraction of filtration, or percentage of the renal plasma flux which is filtered, is of the order of 20%
in man. About 180 liters of glomerular ultrafiltrate are formed by 24 hours, essentially composed of small molecules in solution at concentrations not very different from those of plasma. The existence of non-diffusible negative charges linked to the proteins leads, according to the Donnan equilibrium, to an increase of the diffusible anions and a reduction of the cations in the ultrafiltrate (respectively 5% more or less for the monovalent ions, 9% for phosphates).The passage of the large proteins in the urine is low and depends on their molecular weight and their charge. The filtration ratio is less than 1 for those with a molecular weight greater than 5,000 and the passage is negligible beyond a weight of 70,000. The filtered proteins are essentially reabsorbed downstream in the renal tubule , their concentration in the final urine being normally less than 200 mg / L. Physiological proteinuria appears to be composed equally of proteins of plasma origin (fragments of immunoglobulins and albumin) and of the protein of Tamm-Horsfall, mucoprotein produced by the cells of the cove of Henle.
Glomerular filtration parameters (FG):
These are the filtration coefficient (Kf) and the ultrafiltration pressure (Puf) whose product gives the glomerular filtration rate: DFG = Kf Å ~ Puf. Kf is the product of the coefficient of permeability of the filtration barrier and of the filtration surface. Puf is equal to the algebraic sum of the hydrostatic (P) and oncotic () pressure gradients between the glomerular capillary (CG) and the tubular compartment (T):
The concentration of proteins in the tubular fluid is usually minimal and the resulting oncotic pressure virtually nil; the intratubular hydrostatic pressure is substantially constant. Under normal conditions, therefore, Puf depends essentially on the intraglomerular hydrostatic pressure, regulated by the play of pre- and post-glomerular arteriolar resistances. In normovolemic animals, the Puf is 15.5 mmHg at the entrance of the glomerulus and vanishes before the efferent end when the increase in CG secondary to the loss of the ultrafiltrate cancels δP: this phenomenon is called filtration equilibrium. In the case of volmic expansion, a positive Puf persists at the end of the glomerulus.
In some dysproteinemias, the increase in oncotic pressure in the glomerular capillary can decrease glomerular filtration. Similarly, in the case of ureteric or intratubular obstruction, the increase in intratubular hydrostatic pressure reduces (or cancels) the hydrostatic pressure gradient, therefore the Puf and the glomerular filtration. Finally, the contraction of the mesangial cells induced by numerous vasoactive agents, among which angiotensin II and endothelin, leads to a reduction of the filtration surface, therefore of the Kf and of the glomerular filtration.
Renal self-regulation, tubuloglomerular balance and tubuloglomerular feedback:
The renal blood flow and the glomerular filtration remain constant when the blood pressure changes between 80 and 140 mmHg, permanently ensuring an infusion pressure of 100 mmHg. Glomerular filtration decreases parallel to renal blood flow when the mean arterial pressure is less than 70 mmHg and stops when less than 40-50 mmHg. This phenomenon, called renal self-regulation, involves changes in the related arteriolar resistances. It persists on denervated kidney and involves a myogenic (stretch-reflex) mechanism that originates in the smooth muscle cells of the vascular wall and ensures vasoconstriction of the afferent arteriole in the event of increased perfusion pressure.
The high flow rate of the glomerular filtrate imposes a precise adaptation of the tubular reabsorption mechanisms to the risk of rapid volleic depletion: a 5% imbalance between filtered charge and tubular reabsorption would cause a loss of one third of the blood volume within 24 hours. The balance
glomerulotubular approach describes this adjustment of tubular reabsorption proximal to filtration of the glomerulus of the same nephron. Peritubular and luminal factors contribute to this balance: an increase in filtration is accompanied by an increase in the flow of dissolved substances in the proximal tube and reabsorbed by cotransport systems, and increases the proximal transepithelial osmotic flow favorable to reabsorption of more water. At constant blood flow, an increase in glomerular filtration (by increasing the fraction of filtration) is associated with an increase in the peritubular oncotic pressure which favors proximal tubular reabsorption.
The tubuloglomerular feedback (RTG) represents the modifications of the GFR induced by variations of the flow of tubular fluid of the same nephron. The use of the RTG makes it possible to prevent excessive losses of water and salt by preventing the arrival of an excessive flow of fluid in the distal nephron with limited reabsorption capacities. It is initiated by specialized cells of the macula densa in close contact with the extraglomerular mesangium and the afferent arteriole. The activation of RTG depends on the presence of chlorine and is abolished by furosemide. This suggests the use of a cotransport Na-K-2Cl, identified in these cells, inhibited by this diuretic and whose activity is physiologically limited by a low affinity for chlorine. The effector mechanism of this RTG is a contraction of the smooth muscle cells of the afferent arteriole of the glomerulus as well as those of adjacent nephrons fed by a common arterial branch (phenomenon of cooperativity).
Different systems of coupling between the chlorine signal perceived to the macula densa and the vasomotor response are evoked. The increase in the luminal concentration of NaCl activates RTG and inhibits renin secretion, thus avoiding the mediating role of angiotensin. Adenosine exerts a vasoconstrictor effect on afferent arteriole and inhibits renin secretion; on the other hand dipyridamole increases the luminal concentration of adenosine by decreasing its tubular transport and increases the response of the RTG, while the adenosine receptor antagonists increase the glomerular filtration, suggesting also the intervention of this nucleoside. Thromboxane, whose production is increased in the case of RTG activation, is another candidate: its agonists and antagonists increase and decrease, respectively, the RTG response. Other factors such as endothelin, atrial natriuretic peptide (ANP), nitric oxide (NO) or peritubular physical factors are also able to modulate the RTG response.
Changes in RTG sensitivity are likely to alter glomerular filtration and are most often induced by changes in blood volume and activity of the renin-angiotensin system. Angiotensin II plays a permissive role on RTG, possibly by sensitizing arteriole cells to vasoconstrictor mediators. The administration of angiotensin II antagonists dissociates self-regulation of renal blood flow and glomerular filtration. In the case of volumic expansion, the increase of the NaCl flow at the macula densa activates the RTG and inhibits the secretion of renin and angiotensin, decreasing the sensitivity of the vascular response; the glomerular filtration becomes less dependent on the signal perceived at the macula densa, ultimately allowing the soda excretion by releasing the control of NaCl to the macula.
The importance of changes in glomerular filtration induced by RTG decreases with hypovolemia and decreased renal blood flow. The vasodilatation expected by inhibition of RTG gives place to neurohumoral vasoconstriction. The vasoconstrictive effects exerted on the arteriole afferent by noradrenaline, released locally or circulating and on the arteriole preferably efferent by angiotensin, maintain the Pcg and the perfusion pressure. These vasoconstrictive effects are attenuated by the vasodilating action of the prostaglandins produced by the glomerular cells in response to these same mediators, limiting the risk of renal ischemia. In the case of voloremic depletion, in particular induced by diuretics and even in the absence of renal macrovascular disease, the administration of ACE inhibitors carries a high risk of acute renal failure.
It describes the increase in glomerular filtration observed in different situations and measured during oral protein loading or intravenous administration of amino acids. It involves a renal vasodilation reflected by a decrease in renal vascular resistance, associated with an increase in renal blood flow and in an inconstant way according to the studies, an increase in the fraction of filtration. The mechanism of this response is imperfectly elucidated. Glucagon, whose pancreatic secretion is increased in the postprandial period, increases glomerular filtration; the abolition of this response in pancreatectomized patients or somatostatin, an antagonist of this hormone, suggests a preponderant role of glucagon. The intervention of vasodilating prostaglandins is suggested by some studies showing the attenuation of this response by indomethacin. Endothelium-derived relaxing factor (EDRF) acts as a non-specific mediator of renal vasodilation when the renal functional reserve is involved. The participation of the renin-angiotensinaldosterone system remains controversial.
In the case of renal disease, the amputation of this hyperfiltration capacity does not constitute a preliminary step to the reduction of the glomerular filtration which is usually measured. In different nephropathies or after unilateral nephrectomy, the increase in glomerular filtration induced by a protein load persists to varying degrees. The renal functional reserve is thus not an index of the remaining renal tissue and its measurement can not be used for the evaluation of lesions. It shows a renal capacity of adaptation whose amplitude in absolute value is reduced with the decrease of the glomerular filtration.
Regulation of glomerular filtration:
Many chemical mediators or hormones, produced locally or systemically, cause a decrease in Kf or modulate the activity of pre- or post-glomerular arteriolar sphincters. Vasoconstrictive substances in most cases result in an increase in the production of vasodilating agents (PGE 2 , PGI 2 ). Conversely, vasodilator agents induce the production of vasoconstrictive agents (especially renin).
The stimulation of the intrarenal adrenergic nerve fibers produces a vasoconstriction by stimulation of the α receptors and the decrease of the renal blood flow. Dopamine is vasoconstrictive at high doses but induces a low dose vasodilation blocked by D 2 receptor antagonists.
FUNCTIONAL ORGANIZATION OF THE REAL TUBULA:
The progressive formation of urine results from the succession of phenomena of exchange in contact with specialized epithelia.
Polarity of renal epithelial cells:
apical is characterized by its high viscosity, linked to its richness in proteins and its lipid composition, rich in cholesterol and sphingomyelin. The activity of the proteins of the intestinal or renal epithelial membranes is influenced by changes in lipid content or fluidity observed in certain physiological or pathological situations. For example, the greater phosphate transport activity observed in the superficial cortex compared to the juxtamedullary cortex is associated with a lower apical content of sphingomyelin and cholesterol and greater fluidity.
Metabolic functions:
The oxygen consumption by the kidneys is correlated with the amount of sodium reabsorbed. The required adenosine triphosporic acid (ATP) is 95% produced by the oxidation of substrates extracted from blood: glutamine, lactate, glucose, fatty acids and citrate. A portion of the plasma free fatty acids captured by the kidney is oxidized, the remainder being incorporated into the lipids synthesized by the kidney.
Two thirds of the glucose used by the kidney is completely oxidized to CO 2 ; the remaining third, used by the anaerobic glycolysis of the medulla, is transformed into the lactate following the Cori cycle and reforms glucose in the liver and renal cortex. A very small fraction of the oxidized glucose follows the hexose-phosphate pathway necessary for the synthesis of fatty acids and nucleic acids. Renal glycogen stores are very low and renal glucose production reflects the balance between renal consumption and neoglucogenesis, provided by proximal tubular cells from lactate and pyruvate, α-ketoglutarate and glutamine. The physiological significance of this renal glucose production is reduced and accounts for only 12-25% of total glucose production after a short fast, but increases in the case of prolonged fasting or when glucoseogenesis is decreased. It is stimulated by acidosis, ammoniogenesis and glucocorticoids and is inhibited by insulin.
The tubular catabolism of the filtered peptides and proteins following the absorption and endocytosis phases participates in the turnover of different peptide hormones and avoids the net loss of amino acids that would result from their urine leakage.
Transepithelial transport mechanisms:
The activity of Na-K-ATPase produces an electrochemical gradient very favorable to the entry of sodium into the cell.Secondary transport systems carry out the coupling of this sodium intake to reabsorption (Na-glucose, Na-amino acid or Na-phosphate cotransports) or to the secretion (Na / H, Cl / HCO 3 exchangers) other substances dissolved against sometimes very unfavorable gradients. The transport carried out is limited by the dissipation of the gradient and especially by the number of conveyor units; their saturation and the exceeding of the maximum capacity are translated by the value of Tm measured during functional tests. The different channels identified in the nephron use the electrochemical gradient of the substance transported, the transported quantity depending here on the opening or closing of the channels, whether controlled by a ligand (parathyroid hormone sensitive Ca channel [ PTH]) or by the membrane potential. Other proteins use the hydrolysis of ATP to carry out “active transport” against an unfavorable gradient (H + -ATPase, H + / K + -ATPase). The high permeability to urea of certain segments of the nephron is related to the presence of facilitated diffusion systems recently identified in humans.
Hormonal regulation of cell functions:
The regulation of renal cell functions is provided by signaling pathway factors. A particular case is represented by the receiver
mineralocorticoid of the distal nephron: the affinity of this receptor is identical for the mineralo- and glucocorticoids whose plasma concentrations are nevertheless much higher; the specificity of the mineralocorticoid effect is induced by an enzyme of these cells, 11β-OH-steroid dehydrogenase, which transforms the glucocorticoids into derivatives whose affinity is very low. Glycerhizin contained in licorice inhibits this enzyme and may thus be responsible for some tables of hyperaldosteronism.
The message delivered by a mediator depends on the nature of the ligand and the type of receptor. The redundancy of the hormonal control exerted on the tubular functions, in particular the transport of the cove of Henle, testifies to a combinatorial model of regulation where each hormone no longer exerts a precise single regulating role but where the observed biological effect depends on the sequence and the association of the effects of different hormones on a group of cells.
Endocrine and autocrine functions of the kidney:
Many biologically active substances are synthesized in the kidney and exert a systemic endocrine effect or paracrine control of transport functions, metabolic activities, or kidney cell growth.
Vitamin D:
Synthesis of the active form of vitamin D from hepatic 25 (OH) -vitamin D 3 takes place in the proximal tubular cells under the effect of (OH) D 3 -1α hydroxylase. The activity of this enzyme is increased by PTH and by insulin growth factor 1 (IGF-1), an endocrine factor also produced in the kidney by the collecting duct cells; it is diminished by acidosis and the increase of 1,25 (OH) 2 -vitamin D 3 . Changes in serum calcium have an indirect effect, depending on the secretion of PTH. Variations in the extracellular phosphate concentration in vivo have a growth hormone-dependent effect. These variations also induce a modulation of the synthesis of 1,25 (OH) 2 -vitamin D 3 in vitro, in isolated kidney cells in culture; the mechanism of this direct effect is not known. The effects of vitamin D on tubular transport of calcium and phosphate are controversial.
Erythropoietin:
It is a glycoprotein produced by fibroblastic peritubular interstitial cells in response to variations in the O 2 tissue partial pressure experienced by a “sensor” hemoprotein. Less than 10% of circulating erythropoietin is produced in the liver by some hepatocytes and Ito interstitial cells, also of fibroblastic origin; and astrocytes of the central nervous system (CNS) provide only local secretion of erythropoietin. The erythropoietin receptor is present on the surface of medullary progenitors of erythroblasts, but also megakaryocytes and neurons, suggesting a neurotrophic role.
Erythropoietin controls the production of red blood cells by preventing spontaneous apoptosis of medullary precursors and by inducing erythroid proliferation and maturation.
Endothelin:
known vasoconstrictor peptide. Endothelin is produced in the kidney by endothelial cells and also by mesangial and tubular cells in response to many physical factors (mechanical stress, hypoxia) or hormones (angiotensin II, antidiuretic hormone [ADH], adrenaline, bradykinin, thromboxane A 2 [TXA 2 ], endotoxin, interleukin-1 …). Different receptors have been identified in the internal cortex and medulla, AND binds preferentially ET 1 and determines intense vasoconstriction, and ET B binds the three isoforms ET 1 , ET 2 or ET 3 with the same affinity and responsible, of EDRF, of initial hypotension during the injection of endothelin. The similar distribution of synthetic and receptor sites, very low circulating concentrations and short plasma half-life (<1 min) suggest a mode of autoparacrine action.Endothelin decreases renal blood flow and glomerular filtration; it inhibits renin secretion, stimulates the production of atrial natriuretic factor (ANF) and sodium excretion although it stimulates the production of aldosterone and increases the proximal reabsorption of sodium; it blocks the antidiuretic effect of ADH. Finally, endothelin stimulates the proliferation of mesangial and tubular cells; this effect is potentiated by other mitogenic agents.
Intrarenal renin-angiotensin system (SRA):
Renin, secreted by the cells of the juxtaglomerular apparatus in response to variations in blood volume, is released into the circulation and proteolyses the circulating angiotensinogen of hepatic origin; the converting enzyme converts the released angiotensin I into its active form, angiotensin II.
Angiotensin II exerts vasoconstrictor effects and stimulates adrenal secretion of aldosterone. All the elements necessary for a local production of angiotensin II have been identified in various tissues and in particular in the proximal tubular epithelium, also suggesting an autocrine or paracrine regulation indicated by the high concentration of angiotensin II in the tubular lumen. Angiotensin II stimulates the activity of the Na / H exchanger and the proximal reabsorption of sodium, and increases the intrarenal production of prostaglandins and bradykinins.
Renal kallikrein kinine system (SKKR):
Kallikrein and kalistatin, the kallikrein binding protein, are expressed by the same cells of the distal tube. Kallikrein activates by proteolysis the kininogen of low molecular weight, circulating or produced locally. The synthesis of kallikrein is stimulated by angiotensin II, aldosterone and prostaglandins. The bradykinin and lysyl bradykinin formed are released into the lumen of the distal tube or degraded by kininases, including kininase II produced in the proximal tube and identical to the angiotensin converting enzyme. Kinins are vasodilating and increase renal blood flow but decrease renal resistance and do not alter glomerular filtration. Kinins stimulate the production of prostaglandins that increase renin release and local activity of the renin-angiotensin system; they inhibit the antidiuretic effect of ADH and the reabsorption of sodium chloride in the cortical collecting duct. The effects of kinins are potentiated by ACE inhibitors which prevent their degradation.
Arachidonic acid derivatives, prostaglandins (PG) and autacoids:
The arachidonic acid produced by the liver from linoleic acid and then incorporated into the membrane phospholipids is released during the activation of phospholipase A 2 . The availability of this substrate is the essential limiting factor in the production of prostaglandins, while the nature of the produced metabolites depends on the stimulus and the cell type involved. The essential pathway is the presence of isomerases and reductases to the formation of PGE 2 and PGF 2α , and in the presence of prostacyclin synthase to that of PGI 2 . PGE 2 and PGI 2 are potent vasodilators and stimulate renin production; TXA 2 and endoperoxides are vasoconstrictors. PGE 2 and PGI 2 have a natriuretic effect by inhibiting the transport of sodium in the collecting duct.
However, the main effect of prostaglandins is to modulate the action of certain hormones on renal haemodynamics or tubular transport, and first of all to inhibit the water permeability induced by DHA in PGEs. the collecting channel.Prostaglandins are mostly produced by the cells of the medullary collecting duct and interstitial cells and to a lesser degree in the cortex by glomerular mesangial and arteriolar cells; their synthesis is stimulated by angiotensin II, bradykinin, ADH and calcium and is inhibited by corticosteroids. The half-life of prostaglandins is very short; they are catabolized in ketodericates and excreted in the urine partly in intact form and partly in the form of inactive metabolites.
PAF (platelet activating factor) is produced by the mesangial cells and in the medulla. PAF decreases glomerular filtration by increasing the production of TXA 2 ; it stimulates directly the production of cyclic guanosine monophosphate (cGMP) in Henle’s loop and inhibits the transport of sodium, chlorine and potassium.
Extracellular Nucleotides:
Cyclic adenosine monophosphate (cAMP) formed in the PTH-stimulated proximal tubular cells is partially secreted into the lumen and degraded by adenosine brush border enzymes; it is recaptured in the cell by means of a transporter and allows a resynthesis of cAMP participating in the inhibitory effect of PTH on the transport of phosphate. The luminal cGMP produced in glomerular epithelial cells and those in the broad ascending segment of the cove of Henle after stimulation by the atrial natriuretic peptide inhibits the transport of chlorine in the cove of Henle. The extracellular concentration of ATP is usually very low but may increase after cell lysis or efflux induced by multi-drug resistance (P-glycoprotein).
Paracrine, by binding to P 2 receptors or after degradation to adenosine, extracellular ATP is capable of modulating the activity of calcium channels, potassium channels or other cationic channels. Finally, adenosine, derived from the catabolism of nucleotides by proximal cells, modulates the transport of chlorine and the water permeability induced by ADH after binding respectively to the receptors A 1 and A 2 present along the nephron.
Growth Factors:
The intrarenal production of several growth factors has been demonstrated in various physiological or pathological conditions.
Growth hormone stimulates hepatic production of IGF-1 but also its intrarenal production by the collecting duct cells;an intrarenal production of the different proteins binding IGF-1 (IGF-BP 1 to 6), which modulates its effects, is also demonstrated. IGF-1 stimulates proximal tubular transport of phosphate and 1α-hydroxylase activity; IGF-1 is partly responsible for the stimulatory effect of phosphate depletion on the tubular production of the active form of vitamin D. Epidermal growth factor (EGF) and hepatocyte growth factor (HGF) have mitogenic effects in tubular cells; the effect of EGF is potentiated by angiotensin II. HGF is normally produced by mesangial cells and exerts its effects on tubular motility, proliferation, differentiation and morphogenesis. EGF is normally produced in the distal tube and the collecting duct; its receptors are located in the proximal tube. TGFα (transforming growth factor) and EGF bind to the same receptors with identical affinity. Increased intrarenal expression of EGF, IGF-1 or tubular repair. The interest of therapeutic administration of these growth factors in certain ischemic or toxic kidney diseases is currently being evaluated.
BALANCE OF WATER AND QUALITY OF OSMOLALITY:
Organic water and osmolality:
The total water represents 60% of the body weight and is divided, according to their osmotic content, by two thirds in the intracellular sector and one third in the extracellular sector. The body’s water content is higher in children than in adults; it decreases with age, decreased muscle mass and increased fat mass. Sodium is the main cation in the extracellular sector while its intracellular concentration is kept low by Na-K-ATPase activity; it represents with its anion accompanying more than 90% of the extracellular osmoles. The volume of the extracellular sector depends on the total amount of sodium, while the sodium concentration determines the plasma osmolality, the normal value of which is 290 ± 5 mOsm / kg of water. The water passes freely through the cell membranes. The net flow of water between the intra- and extracellular areas is determined by an osmotic pressure gradient; this flow ceases when the gradient is zero, that is to say when there is an osmotic equilibrium between the compartments. The intra- and extracellular osmolalities are therefore equal and the natramia makes it possible to appreciate the osmolality and the state of hydration of the intracellular sector.
Effective osmolality is usually calculated based on the concentration of sodium and extracellular glucose, ie (140 Å ~ 2) + 5 = 285 mOsm / kg water; it is usually almost exclusively dependent on serum sodium: the plasma concentration of sodium regulates the water balance. Dissociation of the sodium salt in plasma water is in fact incomplete and the “excess” of osmolality calculated by the formula makes it possible to take into account the other extracellular osmoles present (calcium, potassium, magnesium … which represent about 17 mOsm / kg). The increase in urea or presence of alcohol, which freely diffuses through the cell membranes, results in an increase in the total measured osmolality but do not create an osmotic gradient between the intra- and extracellular areas; they do not modify effective extracellular osmolality and intracellular hydration.
Changes in the osmolality of the extracellular medium result in osmotic flows of water and decreased cell volume in the event of hyperosmolality or its increase in hypo-osmolality. These volume variations may be critical for cell function, in particular that of brain cells housed in an inextensible bone cavity. However, variations in extracellular osmolality activate different transport systems, K / Cl exchanger, potassium channels and chlorine in the case of hypo-osmolality, cotransport Na-K-2Cl and Na / H exchanger in case of hyperosmolality, respectively extracellular leakage or on the other hand to retain intracellular dissolved substances and to limit these variations in volume by adapting the amount of intracellular osmoles.
Water balance:
Daily intakes, either in drinking water or in food, vary from one to several liters per day. The sensation of thirst is regulated by the osoreceptors of the hypothalamus but is also directly stimulated by the cutaneous and respiratory losses are not regulated; they increase in the event of hyperthermia or hyperventilation and tracheal intubation, the alveolar air being saturated with water normally recovered by the upper respiratory tract. The kidney assures the zero balance of water and osmoles by adjusting the volume and osmolality of the urine.
Mechanisms of water excretion:
Most of the filtered plasma water is passively and obligatorily reabsorbed into the proximal tube and the descending branch of Henle’s loop following the osmotic gradient created by the activity of the transport systems.
The very high water permeability of these segments is related to the presence of recently identified specialized proteins called “aquaporins” (CHIP-28 or AQP1, AQP3, AQP4) or water channels, insensitive to DHA. The adaptation of the excretion of water to the water charge relates to the volume of fluid entering the distal tube and the collecting tube, usually of the order of 5 to 10% of the glomerular filtrate, ie 15 L / d. The formation of a diluted urine requires three steps: a sufficient glomerular filtrate flow must reach the tubular dilution segment (Henle’s loop); in this segment, dilution of the urine is achieved by reabsorption of sodium and chlorine without water reabsorption; the inhibition of the secretion of ADH allows the maintenance of the hypo-osmolality of the urine.
Dilution segment and corticopapillary osmotic gradient:
The concentration and dilution functions of urine are inseparable: Na and Cl reabsorption in the broad ascending segment of Henle’s loop allows the dilution of the luminal fluid and constitutes the first stage of the accumulation of dissolved substances in the interstitium of the medulla. The medullary countercurrent systems amplify this elementary effect and gradually establish an osmotic pressure gradient up to 1200 mOsm / kg at the tip of the papilla whereas the interstitium of the cortex remains iso-osmotic to the plasma. The spontaneous dissipation of the gradient is limited by the continuous activity of the cotransport Na-K-2Cl and by the low flow circulating in the vasa recta. In the case of osmotic diuresis or treatment with furosemide, the osmotic gradient is abolished due to an increase in the flow in the loop, an increase in the medullary blood flow or, in the case of furosemide, inhibition of cotransport Na-K-2Cl; the urine is then iso-osmotic to the plasma. In the cortical collecting duct, the reabsorption of water determines a concentration of urea; in the medullary collecting duct, ADH increases permeability to urea by facilitated diffusion systems (UT 2 ).The recycling of urea, diffusing from the collecting canal to the interstitium and the fine descending branches of Henle’s loops, allows its accumulation in the medullary where it represents more than half of the osmoles.
Urine concentration and dilution; role of the antidiuretic hormone:
In the absence of ADH, the distal nephron is impermeable to water; nearly 15 liters of fluid normally leaving the dilution segment can then be eliminated, which is demonstrated by the large capacity to adapt to a load of massive ingestion of hypotonic liquids, the absence of simultaneous supply of osmoles may be a limiting factor in the excretion of water and cause the occurrence of rare dilution hyponatremia called “beer drinkers”.
The formation of a hypertonic urine depends on the existence of the medullary concentration gradient and the posthypophyseal secretion of ADH. Beyond an osmotic threshold close to 280 mOsm / kg (set point), the increase in plasma osmolality determines, with a variable sensitivity according to the individuals, a quasi-linear increase in the secretion of ADH. The osmotic threshold for triggering thirst is higher, on the order of 295 mOsm / kg, and corresponds to a maximum stimulation of urine concentration. In the collecting duct, the ADH induces the apical insertion of water channels “aquaporins AQP2” contained in submembrane vesicles by binding to the V2 receptors coupled to the adenylate cyclase; the luminal fluid then equilibrates with the hyperosmotic medullary interstitium which performs water subtraction and the concentration of urine. The physiological importance of AQP2 is demonstrated by the demonstration of mutations of their gene in certain forms of diabetes insipidus.
The stability of plasma osmolality depends on the ability of the kidney to remove water and osmoles independently.This capacity is reflected by the calculation of the clearance of the free water, ie the amount of water to be added (CH 2 O <0) or to remove (CH 2 O> 0) urine to make it iso-osmotic to plasma. The calculation of the clearance of free water of electrolytes (C e H2O ) refers only to osmotically active substances; it does not take into account osmolality related to urea and is a more accurate reflection of the renal response to variations in plasma osmolality.
Osmolar Clarity : C Osm = (U Osm ΔV ) / P Osm
Free water clearance : C H2O = V – C Osm = V [1 – (U Osm / P Osm )]
Clarity of electrolyte free water: C e H2O = V [(U Na + K / P Na ) – 1]
Non-osmotic factors of antidiuretic hormone secretion:
The sensitivity of osmoregulation of ADH secretion increases with age, hypovolemia or angiotensin secretion. The set point can be modified in different physiological (pregnancy) or pathological situations. Cold, nausea, hypoglycemia, pain are non-osmotic stimuli of DHA release. The main factor, however, is the decrease in the volume of blood that triggers, if it is sufficient, a maximum stimulation of ADH secretion. Except in the case of osmotic diuresis, the finding of a high urinary osmolality, that is to say inappropriate in the case of hyponatremia, testifies to a secretion of ADH most often due to a non- osmotic and in the first place a real or effective hypovolemia. Many drugs interfere with the dilution power of urine by stimulating or more rarely by inhibiting the secretion of ADH. Diabetes insipidus induced by lithium has a different mechanism: highly concentrated in the lumen of the collecting duct due to the subtraction of water, lithium penetrates the cells through the sodium channel and diminishes the hydro-osmotic effects of l ADH by reducing the production and cumulative cell accumulation of cAMP; the amiloride inhibiting the sodium channel and the lithium cell input is likely to decrease these manifestations.
SODIUM SUMMARY AND QUALITY OF THE VOLLEY:
The extracellular volume represents 20% of the body weight; it is divided into a plasma sector (3 liters) and an interstitial sector (9 liters). The total amount of exchangeable sodium determines the volume of the extracellular sector.Exchangeable sodium accounts for 70% of total sodium; the remaining 30% is fixed in the bone. The regulation of the extracellular volume depends on the effective volume determined by the contents of the arterial vascular bed, effectively infusing the arterial tissues and baroreceptors of the carotid sinus and the afferent arterioles of the glomeruli.
Sodium intake ranges from 100 to 200 mmol / d, half of the salt added to food (1 g of NaCl brings 17 mmol of sodium).Absorption is almost complete in the small intestine and colon. Extrarenal outflows of sweat (50-80 mmol / L) and fecal are usually low and unregulated.
The kidney alone adapts the sodium balance. In the case of sodium loading and daily intake greater than 340 mmol, the kidney excretes for 2 to 3 days a quantity of sodium lower than the intakes and develops a positive balance of 150 to 300 mmol, corresponding to a weight gain of 1 to 2 kg and an increase of 1 to 2 liters of the extracellular volume. In the case of sodium restriction (5-10 mmol / 24 h), a negative cumulative balance develops, resulting in a loss of 1-2 kg until a new zero-balance situation.
Renal transport and sodium excretion:
The first step in removing sodium is glomerular filtration of plasma sodium, 24,000 mmol of sodium / 24 h (0.12 L / min Å -1,440 min Å ~ 140 mmol / L); the feedstock usually represents less than 1% of the filtered sodium feedstock and therefore eliminates the tubular reabsorption of the remaining 99%.
Tubular reabsorption of Na is closely dependent on the functioning of the Na-K-ATPase which performs an active transport of sodium and feeds the electrochemical gradients necessary for the operation of the various passive transport systems involved along the nephron. In the proximal tube, the low osmotic pressure gradient generated by bicarbonate reabsorption and transport systems (sodium-dependent cotransports and Na / H exchanger) makes it possible, due to the very high water permeability of this segment, to reabsorb two thirds of the glomerular filtrate.Peritubular physical factors (hydrostatic pressure and oncotic pressure) are the main factors controlling the proximal reabsorption of sodium, which is also stimulated by angiotensin II and catecholamines.
The broad ascending branch of the cove of Henle is the seat of the reabsorption of 25% of the filtered sodium; this reabsorption involves a cotransport Na-K-2Cl inhibited by furosemide and bumetanide. In the distal tube, 5% of the filtered sodium is still reabsorbed by a Na-Cl cotransport sensitive to thiazide diuretics and by the coupled operation of two Na / H and Cl / HCO 3 exchangers. In the collecting canal, sodium reabsorption is quantitatively modest but it conditions the adaptation to food intake and the hormonal regulation of the sodium balance: it involves an apical sodium channel stimulated by aldosterone and inhibited by amiloride; the mutation of the β and γ subunits of the canal has recently been identified as responsible for Liddle’s syndrome, which produces a table of primary hyperaldosteronism with arterial hypertension, hypokalaemia and low aldosteroneemia, highly sensitive to amiloride but resistant to inhibitors competitiveness of aldosterone.
Diuretics, mechanisms of action and resistance:
the potential efficacy of diuretics acting upstream of these segments; this adaptation also explains the remarkable synergy of the combination of loop and thiazide diuretics observed in certain situations.
Diuretics inhibiting sodium reabsorption upstream of the distal tube increase sodium throughput at the macula densa and activate the tubuloglomerular reflex. This reduces glomerular filtration and the amount of sodium filtered, and reduces the potential efficacy of the diuretic. The natriuretic effect of acetazolamide, which depends on the inhibition of the proximal reabsorption of bicarbonates, is further limited by the metabolic acidosis secondary to the bicarbonation which it entails. The potency of loop diuretics, furosemide and bumetanide, is partly due to their inhibitory effect on the tubuloglomerular reflex which prevents this mechanism of resistance.
The binding to the albumin of the various diuretics limits their extravascular diffusion and promotes their tubular secretion, thus increasing their effectiveness; spironolactone acts by diffusion to the basolateral pole of the collecting duct cells and then intracellular binding to aldosterone receptors and its effect is less sensitive to a decrease in albuminemia. The natriuretic power of spironolactone is conditioned by the degree of stimulation of aldosterone secretion. It is most pronounced in the case of hydrosodic retention associated with effective hypovolemia (congestive heart failure, ascitic cirrhosis); in the absence of activation of the renin-angiotensin-aldosterone system, its natriuretic power is very low.
Diuretics: side effects
The natriuretic effect of the various diuretics is accompanied by secondary disturbances in the transport of other dissolved substances, varying with their mechanism and site of action. Loop diuretics inhibit potassium reabsorption in Henle’s loop; on the other hand, like all diuretics increasing distal sodium flow, they stimulate potassium secretion in the collecting duct and are responsible for hypokalaemia. Diuretics acting downstream of the distal tube prevent the formation of the negative luminal potential of potassium secretion and are known as potassium sparing.
Apart from acetazolamide, the bicarbonaturic effect of which is responsible for metabolic acidosis, and which is indicated by specific indications, other diuretics are responsible for metabolic alkalosis secondary to volmic contraction and potassium depletion.
Finally, the reduction in tubular sodium reabsorption obtained with all diuretics results in an increase in fluid flow in the medulla and promotes the dissipation of the corticomedullary osmotic gradient, altering the concentration-dilution functions of the urine and favoring the occurrence of hyponatremia. This risk is compounded with the use of thiazide diuretics or the loop, which act directly in the dilution segments.
Regulation of the sodium balance, control of the extracellular volume:
The variations of the effective volume are perceived by the baroreceptors, aortic and arterial aortic and carotid, acting essentially by the modulation of the sympathetic activity and the secretion of ANP; baroreceptors are also present in the afferent arteriole of the glomerulus and form an integral part of the juxtaglomerular apparatus, inducing locoregional and systemic changes in angiotensin II concentrations.
Tubular sodium reabsorption depends mainly on peritubular physical factors in the proximal nephron; the luminal flow of sodium is its principal determinant in the loop of Henle and the distal tube. Although the hormonal regulation of sodium transport is modest in these segments, the predominant role played by aldosterone in the control of the sodium balance depends on its effect in the collecting duct; the physiological importance of natriuretic factors in the regulation of the sodium balance is not known. The redundancy of the control systems makes it possible to compensate for the possible deficiency of one of them. Thus, in the case of primary hyperaldosteronism, the phenomenon of escape to aldosterone accounts for the progressive adaptation of natriuresis and the absence of edema; it is sodium-specific and involves the modification of peritubular hemodynamics, an adaptation of the secretion of ANP and a pressure natriuresis associated with the elevation of arterial pressure. Finally, in the case of primary hyperaldosteronism, the existence of hypokalaemia suggests the persistence of the sodium supply to the distal nephron and the non-respect of the deodorized regime.
The activity of the renin-angiotensin-aldosterone system increases in case of hypovolemia or reduction of sodium intake. The released angiotensin II exerts a direct arterial vasoconstrictor effect and potentiates the release and effect of noradrenaline; it induces the renal synthesis of prostaglandins which stimulate the production of renin and have a vasodilatory effect on afferent and efferent arteriole, limiting the vasoconstrictive effect of angiotensin II and catecholamines. Angiotensin II increases renal sodium reabsorption by its effects on glomerular hemodynamics, its direct tubular effects and the stimulation of aldosterone secretion. The secretion of aldosterone is also directly stimulated by the increase in serum potassium, pituitary corticotropic hormone (ACTH) and acidosis; the stimulating effect of the decline in serum sodium is often masked by associated variations in blood volume. The secretion of aldosterone is inhibited by dopamine and ANP. The interrelations between the renin-angiotensin, prostaglandin and kallikrein-kinin systems have been previously considered.
An inverse correlation is noted between the activity of the renal sympathetic nerves and that of the intrathoracic baroreceptors. The increase in adrenergic activity observed in the case of volume depletion tends to increase the proximal reabsorption of sodium by a direct stimulation α1 of the sodium transport and in a secondary way to the increase of the filtration fraction and to the modification of the l peritubular hemodynamics; by effect β1, it increases renin secretion. This limits the importance of natriuresis induced by the adrenergic increase in blood pressure.
Dopamine exerts a low vasodilator effect on pre- and post-glomerular arterioles, increases renal blood flow and reduces the fraction of filtration; a vasoconstrictor effect dependent on α stimulation appears with high concentrations.Dopamine decreases the proximal reabsorption of sodium by inhibiting the activity of the Na / H exchanger and Na-KATPase.
It induces the local production of prostacyclin and prostaglandins which participate in its vascular and tubular effects.Dopamine is synthesized in the proximal tubule and produces an autoparacrine system involved in the renal control of the sodium balance.
Atrial natriuretic peptide and natriuretic peptides:
The ANP is the first representative of a family of peptides whose mechanism of action is identical and depends on the activation of a particulate guanylate cyclase. The cardiac synthesis of ANP is modulated by variations in hydrosodic inputs. PNA causes vasodilatation of afferent glomerular arteriole and efferent vasoconstriction, increases glomerular filtration and filter fraction. It inhibits the apical sodium channel of the internal medullary collecting canal; its natriuretic effect does not lead to hypokalaemia. ANP decreases the proximal reabsorption of sodium by a direct effect and an indirect effect by stimulating the production of dopamine. Finally, ANP inhibits the release of renin, angiotensin, aldosterone, catecholamines, pro-ANP, cleavage at a different site confers urodilatin resistance to endopeptidase metabolizing ANP. Natriuretic cerebral peptide (BNP) and natriuretic peptide C (CNP) are the products of different genes but have a molecular structure very similar to the ANP and share its mechanism of action; CNP is mainly synthesized in the brain, heart and kidney, and BNP produced in the heart. Their physiological role, like that of the ANP, is not well known. Several receptors of these natriuretic peptides have been identified: receptors B A (GC-A) and B B (GC-B), both formed of membrane guanylate cyclase and different by their extracellular domain, and type C receptors lacking biological activity, predominantly represented and involved in the clearance of the peptide.
Adrenomedullin:
This peptide of 52 amino acids isolated from a pheochromocytoma is physiologically produced in numerous tissues including the kidney. It exerts a natriuretic effect by preventing distal sodium reabsorption; its mechanism of action is not known but is independent of cGMP production and activity of the renin-angiotensin-aldosterone system.
Endogenous Ouabain:
The existence of an endogenous digitalis factor is very probable. Inhibitor of Na-K-ATPase and responsible for a natriuretic effect and an increase in the tone of vascular smooth muscle, this factor whose origin would be adrenal or that would be produced by the CNS is still not identified. On the other hand, in the cove of Henle, the mono-oxygenase pathway produces a metabolite of arachidonic acid which inhibits Na-K-ATPase.
KIDNEY AND ACID-BASE QUALITY:
The normal acid-base equilibrium is defined in the extracellular liquids by a pH of 7.38 to 7.42, a CO 2 partial pressure of 37 to 42 mmHg and a concentration of bicarbonates of 23 to 26 mM; the buffering capacity of this extracellular sector is provided by bicarbonates and proteins. In intracellular fluids, the pH is close to 7, varying according to the intracellular compartments and the metabolic activity of the tissues; the partial pressure of CO 2 is close to that of the plasma and the buffer systems are represented by proteins, bicarbonates, phosphates and hemoglobin in the red blood cells. The buffering capacity of the intra- and extracellular sectors is equivalent.
During metabolic acid loading, extracellular buffering is immediate; the use of the intracellular buffers is delayed by the diffusion time of the H + ions but accounts, due to the greater volume of this sector, of the buffering of two thirds of the acid charge. In the case of respiratory acidosis, carbonic acid can not be buffered by bicarbonates, the combination of H + ions and bicarbonates leading to the regeneration of carbonic acid; the buffering of respiratory acidosis is therefore carried out exclusively by intracellular systems.
Review of H +:
on the other hand. The complete oxidation of fats and carbohydrates produces 10,000 to 15,000 mmol of CO 2 per day, transported to the lung in dissolved or hydrated form (carbonic acid) and eliminated by ventilation without causing acid retention. The metabolism of sulfur and cationic amino acids produces non-carbonic and non-volatile acids; the oxidation of organic anions (citrate and lactate used in gluconeogenesis) and the metabolism of anionic amino acids represent an alkali source. Inorganic acids and non-metabolizable organic acids release an amount of H + ions proportional to the amount of acid and their pK; they are generally fully ionized at plasma pH. The result is a net acid charge of 1 mEq / kg / d, neutralized by buffer systems and eliminated by the renal route. Maintaining the acid-base balance requires the removal of these fixed acids, the restitution of the bicarbonate buffers used and the reabsorption of the filtered bicarbonates (24 mmol / L Å ~ 125 mL / min of glomerular filtration = 4800 mEq / d ).
Tubular reabsorption of bicarbonates:
It is mainly provided in the proximal tube (85%); the rest of the reabsorption takes place between the end of the proximal circumferential tube and the distal circumferential tube beginning (15%) and ends in the distal nephron (1 to 2%). It is coupled to a luminal secretion of H + ions and dependent on the activity of the membrane carbonic anhydrase (AC) present in the proximal tube. The secretion of H + ions is carried out for one third by a vacuolar apical H + -ATPase and for two thirds by an Na / H exchanger. Two isoforms of this interchange were identified in the TCP (proximal bypass tube): NHE-3 (high affinity apical exchanger involved in the transepithelial transport of bicarbonates) and NHE-1 (low affinity basolateral ubiquitous interchange, involved in maintaining of the intracellular pH). Each H +ion secreted in the tubular lumen is buffered with a bicarbonate derived from glomerular filtration. In TCP, the membrane AC allows the rapid dehydration of the carbonic acid formed and the release of a CO 2 molecule that diffuses freely into the tubular cell, whereas downstream in the nephron and in the absence of In the luminal region, the dehydration of the carbonic acid is slow and its accumulation determines a lowering of the pH of the luminal liquid.In cells, ubiquitous cytosolic AC catalyzes the reformation of a bicarbonate from CO 2 and the release of a H + ion, secreted and recycled in turn to the apical pole; basalateral reabsorption of bicarbonate into peritubular and systemic circulation is achieved by secondary transport systems, Na-3HCO 3 in the proximal tube, K / HCO 3 in Henle’s loop and Cl / HCO 3 in the distal nephron.
The intercellular pathway is responsible for a backscattering flow of the bicarbonates from the peritubular compartment to the lumen of the proximal tube. In the case of volume expansion, the increase in the backscatter flux determines a urinary leakage of bicarbonates and a so-called “dilution” acidosis.
Conversely, in the case of volume depletion and activation of the renin angiotensin-aldosterone system, the released angiotensin II stimulates the apical Na / H and basolateral Na-3HCO 3 activity of the proximal tubular cells and increases the proximal reabsorption of the bicarbonates, an “alkalosis of contraction”. The hypochloraemia often associated with metabolic alkalosis perpetuates alkalosis by increasing the reabsorption of bicarbonates in the collecting channel by a Cl / HCO 3 exchanger stimulated by the decrease in the chlorine luminal concentration.
Hypokalemia with potassium depletion also increases the reabsorption of bicarbonates by renal cells; a sodium inlet maintains electroneutrality by compensating for the loss of cations K and borrows the apical Na / H exchanger, stimulating the luminal secretion of H + ions and the reabsorption of HCO 3 in the proximal tube; on the other hand, potassium depletion in the collecting duct increases the reabsorption of K by H + / K + -ATPase and stimulates the luminal secretion of H + .
The increase in CO 2 partial pressure observed in respiratory acidosis increases the reabsorption of bicarbonates; a decrease in PCO 2 has the opposite effect. PTH decreases the tubular reabsorption of bicarbonates by stimulating the production of cAMP in the proximal tube, unlike angiotensin II.
Hypercalcemia stimulates the reabsorption of bicarbonates and the secretion of H + ions; the resulting effect on acid-base balance depends on the etiology and associated factors.
Distal secretion of H +:
The second stage of renal regulation of acid-base equilibrium requires the excretion of the acid charge of 50 to 100 mEq / d which takes place in the distal nephron. After reabsorption of the bicarbonates, the secretion of H + ions takes place in the presence of urinary acceptors of protons: the NH 3 / NH 4+ system, on the one hand, resulting from the metabolism of glutamine in the proximal tube and, on the other hand, the HPO 4- / H 2 PO 4 buffer system.
The secretion of H + ions in the distal nephron is essentially assured by the H + -ATPase of the collector channel type A intercalating cells and to a lesser degree by an Na / H exchanger, the distal electrochemical gradient being favorable to its activity; a H + / K + -ATPase also participates in the secretion of H + , especially in hypokalaemia. The sodium reabsorption carried out by the main cells of the collecting duct creates a negative luminal transepithelial potential difference favorable to the secretion of protons and makes an indirect coupling of the transport of Na and H + ions.The amiloride blocks the sodium channels of the main cells, abolishes the favorable potential difference normally created by sodium reabsorption and determines a defect of excretion of H + ions. The acid secretion by the distal H + -ATPases is small compared to the high proton flux provided by the Na / H exchanger in the proximal H + ion tube; on the other hand, once the reabsorption of the bicarbonates has been completed, it is possible to generate high gradients of H + ions and maximum acidification of the urine to a pH of 4.5. B-type intercellular cells have basolateral H + -ATPase and produce apical secretion of bicarbonates.
An increase in the number and the activity of the intercalary cells A or B is demonstrated respectively in the case of acidosis or metabolic alkalosis.
Titratable acidity:
The excretion of free H + ions is quantitatively negligible, a urinary pH of 4.5 corresponding to a concentration of free H + ions of only 0.04 mEq / L.
The acidification of the urine, however, determines the formation of titratable acidity by shifting the equilibrium of the buffer system HPO 4- / H 2 PO 4- (pK 6.8) towards the formation of H 2 PO 4 . Low acids present in the glomerular filtrate play a minor role in the acceptance of H + ions, depending on the amount and pH of the urine: creatinine (pK 4.97), uric acid pK 5.75). In certain pathological situations, other weak acids are present and may contribute to the constitution of titratable acidity, such as β-hydroxybutyric acid in the case of ketoacidosis (pK 4.8).
Ammoniogenesis and ammoniuria:
Urinary excretion of ammonia accounts for two-thirds of the total excretion of acid. Glutamine synthesized by the liver is its major precursor: it penetrates the proximal tubular cells apically and peritubly to be converted by glutaminase to alpha-acetoglutarate with formation of 2 NH 4+ , added to light by the Na / H exchanger and not very diffusible. In the downstream segment of Henle’s loop, passive water subtraction achieves a concentration of the bicarbonates still present in the free flow from the interstitium into the collecting channel where it fixes the secreted H + ions; the NH 4+formed can not diffuse and is trapped in the acidic urine. Disorganization of the architecture of the medulla, by preventing the medullary recycling of NH 3 , is responsible for certain tubular acidosis.
Hyperkalaemia prevents medullary accumulation of NH 3 by competition of K + transport with NH 4+ borrowing the Na / K (NH 4+ ) / 2Cl necessary for this recycling in the broad ascending branch of the Henle.
Acute net excretion, urinary anion hole (TAU):
The net excretion of acid can be calculated as:
The filtered bicarbonates are usually completely reabsorbed. Titratable acidity is modulable and can only increase by 50%; it is maximal at a pH u equal to 5.5. It depends on the amount of phosphate available in the urine, which is determined by dietary intakes. The regulation of the net excretion of acid is thus based essentially on the adaptation of the tubular production of NH 3 : this is stimulated by acidosis, hypokalaemia, glucocorticoids and PTH and on the contrary inhibited by l alkalosis and hyperkalaemia. The excretion of NH 4+ increases with urine flow.
The extracellular pH is the main regulator of net acid excretion.
Acidosis stimulates the synthesis of NHE-3 in the proximal tube and in the loop of Henle and increases the secretion of H + ions. The apical membrane insertion of H + -ATPase pumps in the proximal tube and the distal nephron also makes it possible to increase the secretion of H + ions.
The quality of the renal response in the case of acidosis can be assessed simply by calculating the TAU: the urine is electroneutral: (Na + K + indosed cations) = (Cl + anions) Na + K – Cl = indices – indosed cations) TAU = Na + K – Cl;normally> 0.
NH 4+ is the main indosed cation of urine; it is eliminated, in order to maintain electroneutrality, accompanied by a chlorine anion. In the case of metabolic acidosis without plasma anionic hole, the adapted renal response is an increase in the production of NH 4 + , hence chloruresis and indosed cations in the urine: TAU negativity then testifies to the extrarenal origin acidosis, for example in the case of digestive loss of bicarbonates while a positive TAU value is oriented towards the tubular origin of acidosis. In the case of metabolic acidosis with an anionic plasma gap (lactic acidosis, diabetic ketoacidosis, salicylate intoxication, etc.), the urinary elimination of the plasma indosed anions positively affects the TAU value independently of NH 4 + excretion. The estimation of ammoniuria then requires the calculation of the urinary osmotic hole, that is to say the difference between the osmolality measured on a urine sample and the calculated osmolality: calculated urinary osmolality = 2 Å ~ (Na + K) + 16.6 urea (g / L) + 5.5 Å-glucose (g / L).
The presence of indoated anions is here taken into account by the accompanying Na or K cations. The calculated urinary osmotic hole depends essentially on the ammonium salts, ie NH 4 + and the accompanying anion.
POTASSIUM BALANCE SHEET:
Potassium is the main cation in the intracellular sector where it accounts for 98% of the exchangeable K of the body.The extracellular sector comprises the remaining 2% of which about 20 mEq is present in the plasma sector and determines the value of serum potassium. Because of a high membrane permeability compared to other ions, the ratio of intra- (150 mEq / L) and extracellular (3.5 to 4.5 mEq / L) potassium concentrations is the main determinant of the resting membrane potential . A small variation in the amount of potassium present in the plasma area determines a significant variation in serum potassium and this ratio, and therefore may be sufficient to modify cellular excitability, particularly cardiac cells.
On the other hand, potassium is also involved in the regulation of cellular synthesis of proteins and glycogen.
The potassium concentration depends on the total amount of potassium and its distribution between intra- and extracellular media. Digestive absorption and the elimination of food potassium (about 50 to 150 mEq / d) vary little;the homeostasis of potassium therefore depends on its renal elimination, whose regulation is slow, placed under the control of aldosterone. The distribution of potassium between the intra- and extracellular areas is influenced by hormonal or physicochemical factors and plays a leading role in the rapid regulation of serum potassium.
Distribution of potassium between intra- and extracellular areas:
Different factors, involved in physiological situations, modulate the transfer of potassium between the intra- and extracellular areas. Postprandial insulin secretion prevents hyperkalaemia which would result from a net addition of the feedstock to the extracellular area; the elimination of the potassium charge is only achieved after a few hours by the kidney. Muscular exercise is accompanied by local hyperkalaemia which exerts a vasodilatory effect and increases blood supply; the intracellular transfer induced by catecholamines limits its importance.
In the case of acidosis, the importance of the transfer depends on the membrane permeability of the anion associated with the H + proton, the associated intracellular passage of negative charges limiting the exit of potassium. Since membrane permeability of chlorine is low compared to other anions, transfer hyperkalaemia is more severe in hyperchloremic acidosis than non-hyperchloraemic metabolic acidosis (with increased anion gap) or respiratory acidosis, which is accompanied by an increase in the intracellular concentration of bicarbonates.
Renal potassium behavior:
The potassium filtered by the glomerulus is reabsorbed to 80% in the proximal tube passively following the water and sodium flux and the weakly positive luminal potential created by the reabsorption of the bicarbonates. The reabsorption is completed downstream in the cove of Henle by the cotransport Na-K-2Cl. Adaptation to urinary excretion of potassium therefore depends on the regulation of secretion and reabsorption flows in the distal nephron; it usually determines a fractional excretion of 15% of the filtered charge.
Potassium secretion is performed by the main cortical distal nephron cells using a K-Cl cotransport system and low-conductance potassium channels, sensitive to ATP and barium. The diffusion of potassium through the potassium channels depends on the number of open channels and the negativity of the luminal potential; this is generated by the sodium reabsorption by the sodium channels present at the luminal pole of the same cells.
Potassium reabsorption is possible in the distal nephron due to H / K-ATPase identified in the collector tube type A intercellular cells and whose activity is stimulated in the case of chronic potassium depletion and inhibited by potassium loading.
The main determinants of distal potassium secretion are aldosterone and serum potassium. The secretion of aldosterone is increased in case of hyperkalaemia even modest and diminished by potassium depletion; aldosterone stimulates distal potassium secretion by increasing the number of open Na and K channels and the activity of Na-K-ATPase.
Hyperkalaemia stimulates distal potassium secretion by the same mechanisms but independently of the effect of aldosterone. On the other hand, a high fluid flow in the distal tube maintains low the luminal concentration of potassium and maintains a favorable gradient to the secretion.
The increase in distal sodium flow induced by proximal, loop or thiazide diuretics stimulates distal sodium reabsorption and promotes the formation of luminal electronegativity favorable to potassium secretion. This voltage-dependent secretion of potassium is inhibited by the amiloride which blocks the sodium channels whose activity is at the origin of the constitution of this difference of potential. Similarly, in the case of primary hyperaldosteronism, the observance of a deodorized diet limits the sodium flow to the distal nephron and prevents the occurrence of hypokalaemia. Finally, the presence of low reabsorbable anions in the distal nephron (massive ketones, bicarbonates, sulphates, carbenicillin) increases luminal electronegativity and increases the tubular secretion of potassium, which can lead to hypokalaemia.
ADH increases distal potassium secretion; this effect compensates for the decrease in the secretion associated with the decrease in the flow of fluid in the antidiuretic situation.
The potassium gradient between the peritubular capillary and the distal tubular fluid (GTTK) is dependent on the effect of aldosterone and can be evaluated simply from a plasma sample and a sample of urine. The concentration of potassium in the peritubular capillary is not very different from serum potassium. The secretion of potassium is normally negligible in the collecting duct and the osmolality of the distal fluid is close to that of the plasma, except in an aqueous diuresis situation. The tubular concentration of potassium at the end of the distal cortical nephron can therefore be calculated by comparing the urinary concentration of potassium with the water reabsorption carried downstream in the collecting duct and evaluated by the ratio of urinary and plasma osmolalities:
In case of hypokalaemia, a GTTK greater than 4 indicates an inappropriate stimulation of the secretion of potassium (by aldosterone in the first place); on the contrary, in case of hyperkalaemia, a value of less than 7 suggests hypoaldosteronism.
Adaptation to changes in potassium inputs:
In the case of potassium depletion, the decrease in serum potassium determines a decrease in aldosterone secretion and intracellular potassium concentration, especially in distal tube cells. The distal potassium secretion is reduced while the H / K-ATPase activity-dependent reabsorption increases, reducing the excretion of potassium to less than 15 mEq / d.
Renal adaptation to increased potassium intake is early.
The increase in serum potassium and aldosterone secretion is accompanied by an increase in Na-K-ATPase activity and K-distal secretion.
Chronic adaptation to hyperkalaemia, as well as renal failure, also involves an increase in K-secretion stimulated by aldosterone, which can then account for up to half of the intakes.
BALANCE OF PHOSPHATE:
Distribution of phosphate in the body:
The total phosphate content of the organism is approximately 1 g / kg by weight, present essentially in the form of organic molecules distributed predominantly in bone (85%) and in the intracellular compartment (14%).
Less than 1% of the phosphate is present in extracellular fluids. Plasma phosphate accounts for less than 0.1% of total phosphate. It is present for about one third in inorganic form as measured by routine phosphatemetering; weakly bound to proteins, it is mostly in ionized form or complexed with calcium and magnesium. The plasma phosphate concentration is in the range of 25 to 45 mg / L (0.85 to 1.45 mmol / L); it varies during the nycthemer and with age and diet. In children, tubular reabsorption of phosphate and phosphatemia are elevated in order to maintain positive the phosphate balance necessary for growth; on the contrary, there is a decrease in renal reabsorption and phosphatemia. Bone phosphate is present as crystals of hydroxyapatite and precipitates of calcium phosphate. In adults, bone accretion and resorption are equal and the resulting net phosphate balance is zero. The normal diet provides about 1 g / d of phosphate, 70% of which is absorbed into the small intestine. This absorption is the result of two components: an active absorption flux dependent on a cotransport bound to sodium and stimulated by vitamin D, a passive flux dependent on the concentration gradient and not regulated.
The phosphate homeostasis depends on the adequacy between the amount of phosphate eliminated in the urine and that absorbed by the gastrointestinal tract, which is usually non-limiting. The filtered phosphate charge depends on the glomerular filtration rate and the amount of ultrafiltrable phosphate, determined by the concentration of free phosphate (not bound to proteins) and its diffusion according to Donnan’s equilibrium. Regulation of the quantity removed in the urine is performed in the proximal tubule by modulating the reabsorbed amount, normally about 85%. This reabsorption depends on active and saturable transport phenomena; it is closely regulated by hormonal and non-hormonal factors. The maximum amount of reabsorbed phosphate relative to the glomerular filtration rate (TmPi / DFG) reflects the phosphate levels above which the phosphate is removed in the urine. This renal phosphate threshold is not a constant value but varies between 2.4 and 4.5 mg / 100 mL (0.77 to 1.44 mmol / L), in particular as a function of dietary intakes.
Cellular mechanisms of phosphate reabsorption:
Tubular reabsorption of phosphate takes place essentially (70%) in the initial bypassed portion of the proximal tubule.It is less important (10%) but very dependent on the control of the parathyroid hormone in its right part. It is negligible in the loop of Henle and weak (<5%) in the distal tubule. The distribution of the cotransport along the nephron is parallel to the reabsorption function; it has recently been confirmed by the use of molecular probes and specific antibodies.
The tubular reabsorption of the phosphate involves a first active transport step at the luminal pole of the cells, in contact with the glomerular ultrafiltrate, towards the intracellular sector. The intracellular phosphate concentration is very high, of the order of 100 mmol / L; that of inorganic phosphate, measured by magnetic resonance, is of the order of 0.6 mmol / L.
The intracellular potential is negative and intracellular phosphate transport, which is the first stage of its tubular reabsorption, is carried out against an unfavorable electrochemical gradient thanks to a sodium coupled membrane cotransport using the favorable energy gradient provided by Na-K- Basolateral ATPase. It is a saturable process, limiting the reabsorption capacity.
By an expression cloning strategy, RNA sequences inducing a sodium-dependent phosphate transport have been identified; the expression of this RNA along the nephron is parallel to the reabsorption function. After its intracellular passage, the phosphate diffuses passively to the basolateral pole of the cell towards the peritubular capillary, following a favorable gradient, by borrowing other transport systems.
Main factors regulating the reabsorption of phosphate by the kidney:
The phosphate richness of the diet is the main regulator of tubular reabsorption. The reabsorption can reach 99% of the filtered charge in a deficiency situation. This adaptation is an intrinsic property of the tubular cells, independent of endocrine factors. PTH increases renal excretion of phosphate whereas acute parathyroidectomy results in an immediate decrease in fractional excretion, less than 1%. Although the stimulatory effects of vitamin D on the active transport of Pi (inorganic phosphate) through the intestinal mucosa are well documented, its renal effects are controversial. Physiologically, phosphate depletion increases the production of 1.25 (OH) 2 -vitamin D 3 . In vivo, this increase is associated with stimulation of IGF-1 production.
Insulin stimulates renal reabsorption of phosphate. The effect of growth hormone is dependent on IGF-1. The physiological effect of calcitonin, vasopressin, glucocorticoids, glucagon, ANP and PTH-related peptide (PTH-rP) is not established. The acidification of the tubular fluid directly inhibits the cotransport. Respiratory alkalosis is associated with a decrease in the fractional excretion of phosphate but also in phosphatemia due to an intracellular transfer of phosphate.
BALANCE OF CALCIUM AND MAGNESIUM:
Distribution and Calcium Intakes:
Bone calcium accounts for more than 99% of the body’s calcium, 25,000 mmol; 100 mmol are rapidly exchangeable and in equilibrium with the ionized calcium contained in the extracellular medium (about 20 mmol). The calcium intakes are of the order of 25 mmol / d (1 g) partially absorbed in the digestive tract. The net absorption of calcium results from an active absorption flux regulated by 1.25 (OH) 2 -vitamin D 3 and a passive flux depending on calcium concentrations in digestive light and interstitium and variant with calcium intakes; at equilibrium, the zero calcium balance is assured by digestive secretion and urinary excretion of calcium absorbed. Adaptation of digestive absorption to intakes depends on active transport and 1,25 (OH) 2-vitamin-D 3 .
Renal Calcium Behavior:
The cytosolic concentration of free calcium is very low, of the order of 50 to basolateral is against the calcium gradient;it involves a Ca-ATPase in the proximal tube, associated with an Na / Ca exchanger in the distal tube. Basolateral calcium channels have been identified; they could be involved in controlling the cytosolic calcium concentration and restoring intracellular stocks.
Plasma calcium is present in albumin-bound form (40%) and in ultrafiltrable, ionized (50%) or complexed form with organic anions (10%, linked to citrates, sulphates or bicarbonates). The filtered calcium load is 250 mmol, of which less than 2% is excreted. In the proximal tube, 60% of the filtered calcium is reabsorbed passively iso-osmotically and according to the electrical gradient secondary to the reabsorption of the bicarbonates; reabsorption continues in pars recta (10%) by an unknown mechanism. Calcium reabsorption in the broad ascending limb of Henle’s loop (20%) depends on the positive luminal potential generated by the Na-K-2Cl cotransport activity; some elements also suggest the existence of active transport stimulated by PTH and independent of the electrical gradient. The hormonal adaptation of the urinary excretion of the filtered calcium takes place in the distal tube; this segment reabsorbs 90% of the calcium reaching it, ie 5 to 10% of the filtered charge. Finally, less than 2% of the filtered calcium passes to the collecting tube and is removed, in ionized or complexed form. The fractional excretion of calcium and the amount of reabsorbed calcium increase with the filtered charge, but in the case of a zero calcium intake there remains a urinary elimination of about 2 mmol / d.
Regulation of tubular calcium transport:
It is the extracellular ionized calcium that determines the renal and digestive behavior of calcium. The regulation of calcemia and that of the calcium balance are most often entangled but can sometimes be dissociated.
PTH is the major regulatory factor in renal calcium transport. The main determinants of its secretion are the concentration of ionized calcium, the amount of parathyroid tissue and 1,25- (OH) 2 -vitamin D 3 . The sensitivity of the secretory response defines the PTH set-point, ie, the ionized calcium concentration established in a dynamic study, which determines a half-maximal secretion of PTH; it is conditioned by the activity of a recently identified membrane protein coupled by a G protein to a phospholipase C and to the effector route, which is directly sensitive to the calcium concentration. Apart from the parathyroid glands, this “receptor” has also been identified in other tissues involved in calcium homeostasis (in the proximal tube, Henle’s loop and distal tube, calcitonin-producing thyroid cells C and osteoclasts) but also in keratinocytes and the CNS. Receptor activity is also modulated physiologically by magnesemia. Finally, mutations of this receptor have been demonstrated in certain pathologies and directly correlated with the observed set point abnormalities (activating mutations and set point decrease in familial benign hypercalcemia, neonatal hyperparathyroidism, polyosteic fibrous dysplasia, mutations inhibitors in some forms of pseudohypoparathyroidism).
The role of 1,25 (OH) 2 -vitamin D 3 is complex. It stimulates digestive absorption and tubular reabsorption of calcium;in the case of low or no calcium intake, it increases osteoclastic osteolysis and maintains the extracellular calcium concentration at the expense of the bone and the net calcium balance.
Effects of diuretics:
Acetazolamide decreases proximal reabsorption but increases the distal bicarbonate flow and stimulates distal calcium transport; the net result is a small increase in calcium excretion. Furosemide inhibits the distal transport of calcium by a complex mechanism: inhibition of cotransport Na-Cl is accompanied by leakage of basolateral chloride following its electrochemical gradient and cellular hyperpolarization; this hyperpolarization stimulates the entry of calcium through an apical calcium channel. In the collecting duct, amiloride inhibits apical sodium intake, decreases the intracellular sodium concentration and stimulates basalateral Na / Ca exchanger activity and calcium reabsorption. Finally, the diuretic-induced volleic depletion is accompanied by hemoconcentration and an increase in the total calcium concentration, without modification of the ionized calcium.
Magnesium:
Magnesium, another divalent cation, is mainly stored in bone tissue (65%) and intracellular (34%), where it is not exchangeable; Only 1% is present in the extracellular sector. Digestive absorption is passive and depends on dietary intakes. Only 70% of the plasma magnesium is filtered by the glomeruli, the rest being bound to the proteins.
The fractional excretion of magnesium is close to 3%; like tubular reabsorption, it increases with the intakes and the filtered charge. The tubular transport of magnesium occurs mainly in the cove of Henle, of which it represents a marker of activity. Its reabsorption is passive by the paracellular route and follows the electric gradient created by the activity of Na-K-2Cl; it is not very sensitive to calciotropic hormones. Below a magnesemia threshold value of 0.5 to 0.6 mmol / L, urinary excretion becomes zero; in the case of increased intakes, as for calcium, fractional excretion increases as well as tubular reabsorption of magnesium.
BALANCE OF GLUCOSE, AMINO ACIDS AND ORGANIC ACIDS:
Glomerular filtration produces 150 to 180 liters of ultrafiltrate per day; the reabsorption of the essential nutrients they contain takes place in the initial part of the proximal tube while the secretion of organic anions and cations takes place a little downstream. The reabsorption of glucose and amino acids is coupled to that of sodium and represents a positive charge transfer which generates a negative luminal potential, in turn favoring the reabsorption of the bicarbonates.
Glucose:
The glucose is freely filtered by the glomeruli and then almost completely reabsorbed by the sodium-dependent cotransport systems present in the apical membrane of the proximal tube, mostly in its initial part bypassed. Two carriers were identified, both inhibited by phlorizine, SGLT-1 and SGLT-2; they have a high affinity – low capacity and low affinity – high capacity, respectively. SGLT-2 is present in the initial part of the proximal tube while SGLT-1, also present in the intestinal epithelium, is located further downstream. The limiting step of glucose transport is that of the passage of the apical membrane, against an unfavorable concentration gradient; the coupling to the sodium inlet, which follows the very favorable electrochemical gradient maintained by the activity of the Na-K-ATPase, makes it possible to overcome this obstacle and to achieve an intracellular accumulation of glucose.
After its apical passage, the reabsorption of glucose is completed by its GLUT-1 and GLUT-2. The occupation of all transport sites defines the maximum reabsorption capacity (Tm), ie the amount of glucose present in the ultrafiltrate beyond which all of the filtered glucose is removed in the urine.
In humans, this value is close to 360 mg / min (2 mmol / min) or about 3.3 g / L (18.4 mmol / L) of glomerular filtrate.
Amino acids:
The total plasma concentration of the amino acids is of the order of 10 mmol / L. They are freely filtered by the glomeruli. The tubular reabsorption of the amino acids depends on a first apical limiting step of intracellular passage coupled to that of sodium, by a transporter which is also stereospecific and saturable. Depending on the amino acid involved, the basolateral step may be dependent on a cotransport system coupled to sodium. Several families of carriers are identified and allow the transport of different classes of amino acids. The reabsorption of an amino acid can be inhibited competitively or not by other amino acids.
The appearance of an aminoaciduria can therefore reflect the increase in the plasma concentration of an amino acid with saturation of the specific transporter or the lack of tubular reabsorption of an amino acid whose plasma concentration is normal.
Anions and organic cations, uric acid:
The secretion of organic acids participates, along with the glomerular filtration, in their urinary excretion. Plasma para-amino-hippuric acid (PAH) is an organic acid whose tubular secretion conditions renal extraction; it is limited by a maximum secretion capacity. The transport is carried out in two steps: active entry of the PAH at the basolateral pole and apical passive exit following the concentration gradient. When the plasma concentration of PAH is low, extraction is complete when it first passes through the renal circulation; the measurement of PAH clearance then allows the calculation of renal blood flow. Different systems of transport of endogenous organic anions (bile salts, cAMP, fatty acids, prostaglandins) or exogenous (numerous drugs) are identified in the proximal tube; their renal transport follows that of the HAP and is sensitive to probenecid. The less well known secretion and reabsorption processes can vary from one molecule to another.
Organic cation transport systems also allow proximal tubular secretion and the elimination of endogenous and exogenous cations.
The inhibitory effect of probenecid on the transport of organic acids is used to decrease the excretion of certain drugs (penicillins, certain cephalosporins). A small fraction of creatinine is normally eliminated by tubular secretion; this share increases relatively in the case of a decrease in glomerular filtration. Under these conditions, cimetidine or trimethoprim, which diminish tubular transport of creatinine, may result in elevation of plasma creatinine and apparent aggravation of renal insufficiency without, however, decreasing glomerular filtration.
Uric acid is derived from the metabolism of endogenous purine bases and brought by the diet. It is a weak acid predominantly present as urate at plasma pH. It is little related to proteins and its glomerular filtration is free. The fractional excretion of uric acid is 6-12%.
Its renal behavior, which is entirely determined in the proximal tube, results from the reabsorption of almost all of the filtered uric acid in the initial contour, followed by tubular secretion of about half the filtered charge and then reabsorption of a portion of the uric acid secreted in the terminal portion of the proximal nephron. The reabsorption of uric acid depends on an apical system of anionic exchanger operating in parallel with an Na / H exchanger which generates the favorable pH gradient. The basolateral step of reabsorption of uric acid is passive; The diffusion of uric acid, particularly the reabsorption stage, is very dependent on the volumic state. In the case of hypovolemia, the increase in proximal reabsorption leads to an increase in the tubular concentration of uric acid and promotes its passive reabsorption, while the secretion of angiotensin
It stimulates the Na / H exchanger and the reabsorption of uric acid depending on it.
The increase in uricemia is therefore a very early marker of a decrease in blood volume.
KIDNEY AND AGING:
Aging is marked by a gradual decrease in the cortical mass of the kidneys and an increase in the number of sclerous glomeruli (1 to 2% between 30 and 50 years to more than 30% after 80 years). Glomerular senescence may be accompanied by the obliteration of the corresponding pre-glomerular arteriole, with the diminution of the cortical blood flow, or the development of a shunt between the afferent and efferent arteriole. Age also associates with intrarenal vascular alterations independent of arterial hypertension; the renal blood flow is relatively reduced compared to the cardiac output, which is preserved.
The progressive decrease in renal blood flow is parallel to the increase in renal vascular resistance, particularly in the efferent arteriole. This results in an increase in the filtration fraction, especially since the superficial nephrons, which have the lowest filtration fraction, are the most affected by these alterations.
Glomerular filtration decreased with age, on average 0.75 mL / min / year, although decreased muscle mass most often masked a rise in plasma creatinine. Renal filtration reserve is also decreased. The decrease in renal blood flow predisposes to ischemic or toxic lesions, and in particular requires the adaptation of the dosage of drugs in the elderly.However, epidemiological studies have revealed groups of individuals whose glomerular filtration remains constant with aging, showing the non-inevitable nature of this decrease, and suggesting the role of associated factors in this process, in particular food, metabolic , or hemodynamics.
If the balance of water and sodium and of the different electrolytes are not changed in a normal situation, the mechanisms of adaptation to changes in supply become insufficient. The ability of the elderly kidney to conserve sodium in the event of sodium depletion is impaired; this may be related to a decrease in the reabsorption capacity of the distal tube or to a relative increase in medullary blood flow. Plasma renin activity is decreased by 30-50% in the elderly although the renin level remains normal. The decrease in glomerular filtration reflects the decrease in the elimination capacity of a soda load.
The dilution capacity of the urine diminishes with age, preceding the drop in glomerular filtration; the minimum urinary osmolality increases from 50 mOsm / kg to more than 92 mOsm / kg, while the free water clearance decreases from 16 mL / min to 5.9 mL / min. The concentration capacity of the urine of the elderly kidney also decreases, although the secretion of ADH in response to an increase in plasma osmolality is retained or increased. A decrease in the production of cAMP by DHA is evident in the rat collecting channel, without the number and affinity of the receptors being altered, suggesting a post-receptor mechanism. Finally, the decrease in transport in the loop and the relative increase in medullary blood flow decrease the hypertonicity of the medulla and favor the washing of the corticomedullary gradient.
If blood pH, bicarbonateemia and basal acid excretion are unchanged, there is a delay in the elimination of an acid charge associated with a defect in ammonia. The activity of the Na / H exchanger of the proximal tube is, however, preserved.
Aging is accompanied by a decrease in the total pool of K + , in particular the exchangeable pool. The kaliuretic response to a food potassium loading is decreased. Renal acid deficiency and hypoactivity of the renin-angiotensin-aldosterone system may explain the incidence of tubular acidosis type IV.
Intestinal absorption of calcium and phosphate decreases in parallel with decreased 1-alpha-hydroxylase activity in the proximal tube and thus in the production of calcitriol. The ability to adapt intestinal or renal absorption to a dietary restriction of phosphates is impaired. The tubular reabsorption of phosphate is decreased, in direct relation with an increase in the apical cell membrane content of cholesterol, sphingomyelin and saturated fatty acids, which decreases its fluidity and the activity of cotransport sodium phosphate.
In contrast, tubular reabsorption of calcium is not altered.