 Introduction:
Introduction:
Rheumatoid purpura or Schönlein-Henoch purpura is a small-vessel systemic vasculitis related to predominant immunoglobulin A (IgA) immune deposits. This definition was adopted in Chapel Hill in 1994 for the classification of angiitis which include rheumatoid purpura. For this vasculitis, the presence of IgA deposits is required in the small-sized vessels of the skin, intestine or kidney (glomerulus).Rheumatoid purpura is characterized by the association of cutaneous, articular and gastrointestinal signs, which can occur by successive thrusts. Renal involvement sometimes associates with these signs. The frequency of this attack is extremely variable according to the series. More rarely, other organs such as lung, heart or nervous system may be involved.
The prognosis of the disease in the short term depends on the severity of the digestive disorder but in the long term it is dependent on the renal impairment. Here again, the prognosis remains controversial despite recent publications of pediatric and adult series showing the existence of chronic renal insufficiency, sometimes more than 10 years after the first outbreak.
Epidemiology:
Rheumatoid purpura can occur at any age (5 months to 89 years) but mainly affects children between 3 and 15 years of age. In children, the annual incidence of this disease is in the order of 15 to 20.5 cases per 100,000 children per year. It is much more rare in adults, where the incidence is more like 0.1 per 100 000 adults, the ratio of children to adults would thus vary from 16 to 22. Adult rheumatoid purpura differs from that of the child by its incidence and the severity of clinical manifestations.
In children as in adults, the disease is more frequent in male patients (sex ratio: 1.5). The disease appears to be more frequent in the winter, but the incidence can vary widely from one year to the next.
Rheumatoid purpura is reported in all countries of the world, but its distribution is variable. It appears more common in Japan, South-East Asia, Europe and Australia than in North America and South Africa. It is observed in all ethnic groups but is more rare in black subjects.
Physiopathology:
As far as his physiopathology is concerned, little progress has been made. The abnormal response of an immature immune system to external antigenic aggression has been the most frequently assumed hypothesis.
Indeed, some of these patients have abnormalities of the immune system, focusing on IgA, which plays a central role during rheumatoid purpura, responding to an antigen presented by the immune cells of the mucous membranes. There was an increase in serum IgA levels, unbalanced in favor of IgA1 subclasses, circulation of immune complexes composed of IgA, abnormalities of IgA glycosylation and an increase in the number of circulating B lymphocytes bearing IgA membranes and capable in vitro of producing an excess of IgA.
Moreover, the disease is often preceded by an infection of the otorhinolaryngological (ENT) or respiratory sphere (streptococcus, adenovirus, parvovirus, Mycoplasma pneumoniae …) or a medicinal, toxic and alimentary intake, especially in children . It has also been associated with other viral infections such as parvovirus B19, Epstein-Barr virus (EBV), cytomegalovirus (CMV), or human immunodeficiency virus (HIV). More recently, adults have been shown to be associated with certain cancers, particularly mucosal epithelia, which are more frequent in alcoholotabagic intoxication (tumors in the upper and lower respiratory tract), without a true paraneoplastic syndrome can be affirmed.Finally, the association of rheumatoid purpura and familial Mediterranean fever, particularly in children with the M694V mutation of the FMF gene, is frequent, estimated at 5%. One study proposes the systematic search for this mutation, in populations at risk, when the rheumatoid purpura occurs before the diagnosis of FMF.
Clinical Manifestations:
Diagnostic criteria:
The diagnosis of rheumatoid purpura is based on an extremely evocative combination of clinical signs. It is the association of a cutaneous vascular purpura, articular manifestations, digestive and renal. There is no specific biological sign of the disease. The serum IgA level is high in 60% of cases but this is by no means a formal argument for asserting the diagnosis. Skin histology (leukocytoclastic vasculitis) and renal (proliferative endocapillary glomerulonephritis) associated with the presence of IgA deposits in these tissues may be useful, especially in adults.In 1990, the American College of Rheumatology proposed the following criteria to distinguish rheumatoid purpura from other vasculitis: age less than 20 years, vascular purpura, acute abdominal pain or cutaneous leucocytoclastic vasculitis. The presence of at least two of these signs has a sensitivity of 87.1% and a specificity of 87.7%. In 1994, the presence of IgA deposits in the small cutaneous, intestinal or renal vessels was added as a mandatory criterion during the Chapel Hill consensus conference. The most recent criteria are proposed by Helander et al. : in a patient with leukocytoclastic vasculitis the diagnosis of rheumatoid purpura can be made if at least three of the following criteria are present: IgA dermal vascular deposits, age less than 20 years, gastrointestinal involvement (abdominal pain or blood in stool), recent infection of the upper airways and presence of mesangial nephropathy with or without IgA deposits at the biopsy. These two classifications are very useful for making the diagnosis of the disease in children but are not adapted to the adult, the age of less than 20 being one of the main criteria.
General signs:
Fever, usually low, is present in half of the patients. In children, a severe nephrotic syndrome can be complicated by a frank alteration of the general state with significant loss of weight. An inflammatory biological syndrome is common, but generally moderate.
Signs of skin:
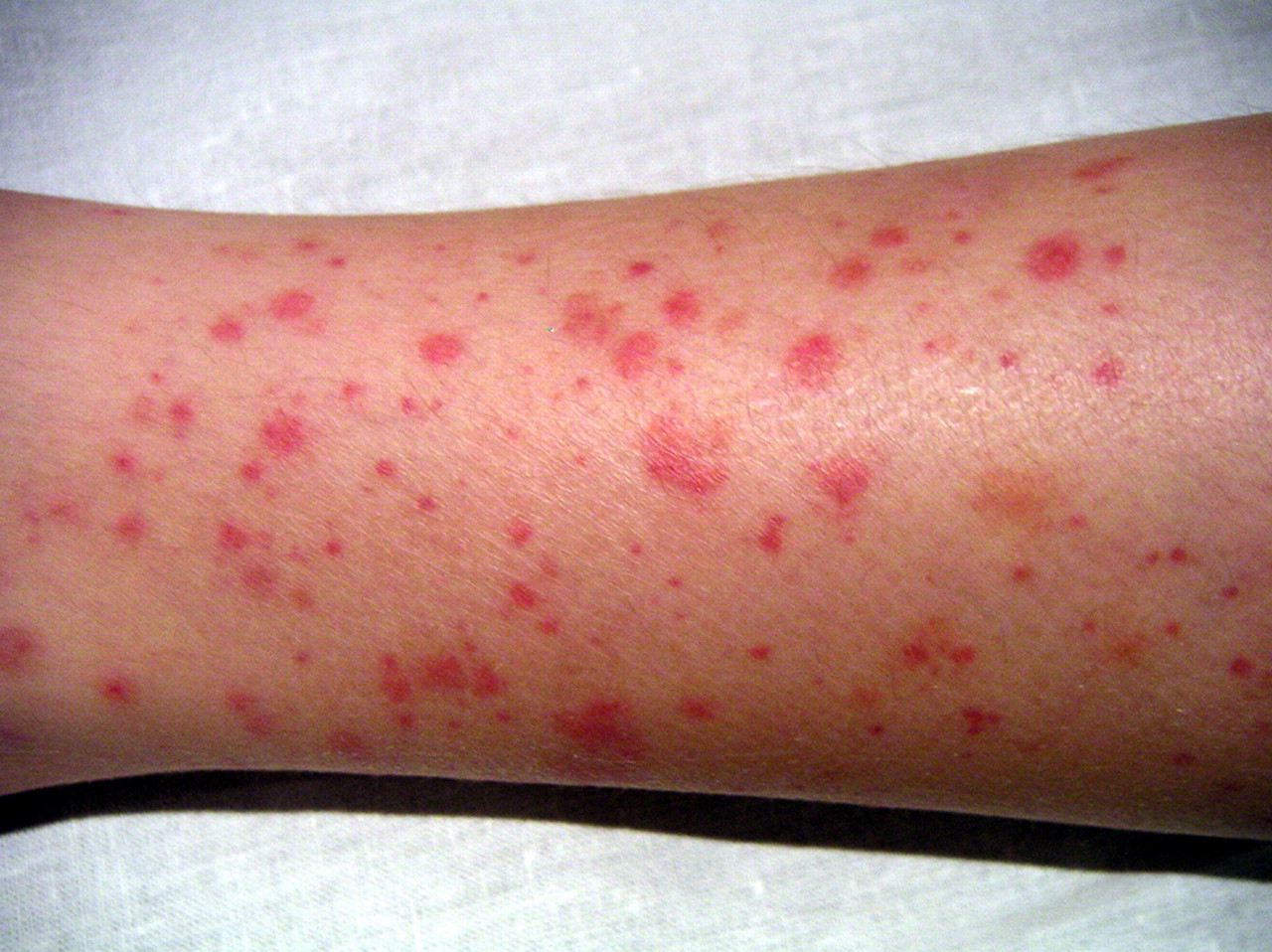
Dermal involvement is almost constant and inaugurates the clinical picture in more than two thirds of the cases. It often begins with urticarial lesions but the characteristic lesion is a vascular purpura. It is symmetrical, not pruriginous, predominates at the pressure zones and at the ends, especially around the ankles and on the buttocks but can extend to the entire integument. It generally spares the trunk, abdomen, face and scalp. Facial involvement is only observed in very young children. The primary lesion is usually an infiltrated petechial purpura, which can conflate to form macules, or even bruises with sometimes cockade aspects. The purpuric lesions are sometimes very discreet, the size of a pinhead. In adults, it is complicated by necrosis or hemorrhagic bullae in 35% of cases, which is exceptional in children. There may be only one rash of rapid resolution or several successive thrusts. Frequent recurrences may however leave a brownish dyschromia or whitish scars in case of necrotic purpura.
The cutaneous skin biopsy of a recent lesion typically shows leukocytoclastic vasculitis of the dermal vessels with fibrinoid necrosis and perivascular infiltrate (capillaries and postcapillary venules) made of neutrophils and mononuclear cells whose nuclei are pycnotic and fragmented (leucocytoclasia) . Immunofluorescence, made on recent lesions (<24 hrs) and infiltrated, shows granular deposits in the walls of the damaged vessels, which shape the vessel wall, polyclonal IgA, C3 and fibrin / fibrinogen. IgM and, more rarely, IgG deposits may be present in the vessel wall.After 24 hours, the immunofluorescence study is often negative.
Signs of joints:
Joint manifestations are present in two thirds of cases and inaugural in one third of the cases. In adults, it is most often arthralgia than arthritis. They result in oligoarthralgia of inflammatory schedule affecting mainly the ankles and the knees. Joint damage is fixed, symmetrical, variable in intensity, often rapidly resolving. One or more joints are affected, simultaneously or successively. Pain may be associated with periarticular swelling, usually associated with a synovitis which, by definition, never destroys the joint. The radiology of the joint may show a periarticular edema, without modification of the interarticular space.
Abdominal Signs:
Digestive manifestations are frequent, variable according to the series (on average 66% in children and 50% in adults), inaugural in 10% of cases. These are moderate pain of the colonic type but may be severe, leading to laparotomy.They are often accompanied by digestive disorders such as nausea or vomiting.
They may associate with occult digestive hemorrhage (presence of blood in the stool with the band) but sometimes serious (hematemesis or melena), engaging the vital prognosis. The digestive endoscopy then finds an erythema of the mucosa with petechial purpura, erosions, or even true beaches of necrosis of the digestive wall. In children, intestinal intussusception is a classic complication of digestive involvement, accounting for 80% of the surgical complications of rheumatoid purpura in children. The digestive histology is superimposed on that of the skin and the examination in immunofluorescence confirms the diagnosis in the presence of deposits of IgA of the vessels of the submucosa.
Other extrarenal manifestations:
The other manifestations are much more rare. Neurological manifestations include headache, convulsions, paresis, and even coma. More exceptionally, orchiepididymitis (most often in young boys), urethritis, pancreatitis, parotitis, myositis, episcleritis, pulmonary haemorrhage (severe) and myocarditis are described.
Renal involvement:
Renal involvement occurs in 20 to 54% of cases of rheumatoid purpura in children and 45 to 85% in adults.
The incidence varies according to the series, depending on the mode of recruitment of the patients and the diagnostic criteria used to confirm the renal impairment. Of all the glomerulonephritis in children, rheumatoid purpura accounts for 10-15% of cases and 2.5% of end-stage renal disease. In adults, renal involvement of rheumatoid purpura would represent only 0.6 to 2% of nephropathies.
Renal involvement usually occurs during the first month of the disease, but urinary abnormalities can be observed after several months, sometimes during a new outbreak of cutaneous purpura.
Hematuria, most often microscopic, is the earliest sign. To this haematuria can associate other evocative signs of glomerulonephritis: proteinuria of variable flow, which can be nephrotic, and renal insufficiency. In adults, renal involvement is not only more frequent but also more severe. The presence of renal insufficiency at the time of diagnosis is exceptional in children, whereas its incidence can reach 32% in adults. High blood pressure may associate with these signs or be isolated.
The nephropathy of rheumatoid purpura is a glomerular nephropathy with deposits of IgA comparable to that observed during Berger’s disease. Only the clinical context makes it possible to distinguish them. The lesions observed during rheumatoid purpura would be more inflammatory and necrotic.
This can be explained by the fact that, unlike Berger’s disease, biopsy is usually performed in the acute phase of the disease.
The immunofluorescence study confirms the diagnosis of glomerulonephritis with IgA deposits. Mesangial deposits are present in all glomeruli, their abundance is variable and they can even extend along the walls of the glomerular capillaries, or even of the vascular pole. These deposits of IgA (essentially IgA1 lambda more intense than kappa) consistently associate deposits of C3, sometimes of IgG (40% of the cases) and more rarely of IgM (20% of the cases). Mesangial deposits of IgA are observed in sclerotic glomeruli, making it possible to carry out a retrospective diagnosis. The necrotic lesions and the croissants fix the antifibrinogenic serum.
The association of fibrinogen and mesangial IgA, even in the absence of fibrinoid necrosis, is suggestive of nephropathy of rheumatoid purpura rather than of Berger’s disease.
Optical microscopic examination shows a great diversity of type and severity of glomerular lesions. Many classifications have been proposed, none of which are unanimously accepted. All are based on the degree of endocapillary proliferation, the number of croissants and the extent of glomerular sclerosis. The classification most commonly used in children is that of the International Study of Kidney Disease in Childhood (ISKDC) modified by Heaton in 1977. We proposed for the adult a classification for which there is a strong anatomoclinic correlation . It distinguishes:
• mesangiopathic glomerulonephritis, characterized by the presence of normal or subnormal glomeruli with a discreet thickening of the mesangial axis;
• segmental and focal glomerulonephritis characterized by the presence of segmental lesions affecting less than 50% of the glomeruli. These segmental lesions may have different aspects. It may be focal mesangial proliferation with matrix augmentation, sometimes accompanied by segmental necrosis of the flocculus and always adherent to either a segmental epithelial crescent or a fibrous or fibrocellular synechia from the flocculus to the capsule of Bowman.Sometimes these lesions of true lesions of segmental and focal hyalinosis of the flocculus are added to these lesions;
• diffuse endocapillary proliferative glomerulonephritis, characterized by the association of mesangial hypercellularity with thickening of the mesangial matrix. Classes IIIa and IIIb are distinguished by the intensity of endocapillary proliferation and / or its association with extracapillary proliferation;
• endocapillary and extracapillary glomerulonephritis, characterized by the presence of crescents, affecting more than 50% of the glomeruli, which may be circumferential, associated with diffuse endocapillary proliferative glomerulonephritis;
• and finally, the fibrous, terminal kidney. In children, the most common lesion is segmental and focal glomerulonephritis; whereas in adults the most common lesion is diffuse endocapillary proliferative glomerulonephritis (class III). Endo- and extracapillary proliferation is rare, observed in less than 10% of cases. We can also observe less specific lesions such as fibrinoid necrosis, which occurs mainly in the acute phase.
Tubulo-interstitial lesions are frequently associated with glomerular lesions and are strongly correlated with the presence of renal insufficiency. Hematic cylinders are readily available. Vascular lesions are also common, even in young adults. This is medially-intimal hypertrophy of the small arteries and arteriolosclerosis, less often arteriosclerosis or fibrotic endarteritis of the interlobular arteries and exceptionally of necrotizing angiitis (with IgA deposits) more or less inflammatory disease affecting the muscular arteries.
In electron microscopy, in addition to visible lesions in light microscopy, there are electrondense deposits in the mesangium, sometimes with subendothelial deposits and rarely subepithelial deposits which, when voluminous, take up appearance of humps.
Evolution:
In children, rheumatoid purpura is most often manifested by a single thrust. In adults, 22% of patients will have multiple outbreaks and 33% of adults will become chronic.
The vital risk is primarily related to digestive involvement, when it is complicated by perforation or uncontrolled gastrointestinal haemorrhage. These complications, which are more frequent in children, are nonetheless exceptional.Pulmonary involvement (intra-alveolar hemorrhage) is very rare but often fatal. The long-term prognosis depends mainly on the course of renal disease.
The risk of progression to end-stage renal disease requiring dialysis in children is variable, ranging from 2.5% to 25%, but on average around 8%. In Europe, 3% of children are dialysed because of a rheumatoid purpura. Two studies concerning children followed more than 20 years after the diagnosis of rheumatoid purpura give a sufficient recoil with regard to this pathology. They emphasize the need for prolonged nephrological follow-up. Indeed, even children considered in remission 10 years after diagnosis developed 20 years later arterial hypertension or chronic renal insufficiency. This monitoring is essential in cases of pregnancy occurring in patients with a history of rheumatoid purpura. One third of pregnancies in the first series and 70% in the second series are complicated by gravid or toxicemic hypertension and up to 16% fetal death.
In adults, the risk of developing chronic renal insufficiency is common, ranging from 8 to 68%.
These differences in outcomes are due to many factors: small series of patients, large discrepancies in age from one study to another, pattern of recruitment (with or without renal involvement) and decline in studies. Among recent studies including a sufficient number of patients, all with renal impairment justifying a renal biopsy, an Italian study finds an incidence of end-stage renal failure of 17% in the 97 adults followed on average 4.9 years. Our retrospective study found an incidence of terminal kidney failure of 18.6% among the 250 adults followed on average 5.2 years.
The results of the published series, both in pediatrics and in adults, during rheumatoid purpura, renal involvement in children is more rare and better prognosis. However, again, it is important to underline the great disparity of these studies, both in the number of patients included, in the number of patients with renal involvement, confirmed or not by a renal biopsy, but also in follow-up, sometimes very short and especially with regard to the treatments received.
Prognostic criteria:
Most studies, whether in children or adults, agree that there is no correlation between the presentation or intensity of extrarenal signs and the histology or progression of l Renal involvement.
In children, there is a fairly good correlation between nephrologic clinical presentation, renal histological examination and renal progression. The presence of a nephrotic syndrome with renal insufficiency or a nephritic syndrome is more often associated with histological stages IV and V of the ISKDC classification and is an important risk factor for the progression to renal insufficiency. Isolated haematuria and / or moderate proteinuria less than 1 g / l is, on the other hand, more often associated with Stages I to III of the ISKDC classification and generally progresses towards remission. The three main criteria retained by most authors remain, the presence of a nephrotic syndrome and / or renal failure at diagnosis and the presence on the renal biopsy of crescents occupying more than 50% of the urinary chamber and more than 50% of the glomeruli. Other factors have been correlated with lower renal survival, found in individual studies: age of child between 5 and 10 years, persistence of cutaneous purpura, presence of abdominal purpura, severe abdominal symptomatology, decreased factor XIII activity.
In adults, these are often small series and no correlation can be made. The Italian study finds, as a factor of poor renal prognosis, proteinuria with a diagnosis of more than 1.5 g / d, as well as the presence of renal insufficiency and arterial hypertension. We also showed, in multivariate analysis, that the existence of renal insufficiency and proteinuria greater than 1 g / d were prognostic factors of severe renal failure in the long term. There are also histological prognostic factors: glomerular fibrinoid necrosis, global glomerular sclerosis and interstitial fibrosis, the prognostic value of which is now accepted for many glomerulopathies, and in particular for Berger’s disease. Contrary to what is observed in children, the presence of extracapillary proliferation does not seem to influence the prognosis.
Treatment:
Symptomatic treatment:
Rest in bed limits the extension of cutaneous purpura but does not influence the development of digestive or renal involvement.
It should be limited to patients with joint pain such that mobilization is hardly feasible. In the first intention, simple analgesics are proposed. Non-steroidal anti-inflammatory drugs (NSAIDs) are of course contraindicated in patients with impaired digestive or renal function.
Finally, nephroprotection measures are recommended in any patient with renal impairment. Conversion enzyme inhibitors, if required with angiotensin II receptor antagonists, should be used as first-line therapy for optimal control of blood pressure and proteinuria output. Specialized monitoring is necessary as long as clinical or biological abnormalities persist.
Then, if there are no urinary abnormalities, normal kidney function and controlled blood pressure, an annual follow-up is recommended.
These symptomatic measures are usually sufficient in most patients. More specific treatments have been proposed for patients with clinical form of concern.
Corticosteroids:
Corticosteroids are effective in reducing abdominal and joint pain, such as common analgesics. They have not been shown to be effective in preventing digestive complications but are generally used in severe cases with strict medical and surgical supervision. The authors agree that they are ineffective on cutaneous disease. To prevent kidney complications, the results of studies using steroids alone are contradictory. Two studies show their beneficial effect, while the others do not find any effect of the treatment.
As for the curative treatment, here also the studies are of difficult interpretation since the most often retrospective and that the treated patients generally present the most severe form. Steroids used intravenously appear to be effective in children.
Immunosuppressive treatments:
More aggressive treatments, involving steroids and cyclophosphamide or azathioprine, appear to be more effective, but here too the results of these studies are difficult to interpret since they include only a few patients and are retrospective or without a control group. In practice, immunosuppressive treatments are used in severe forms with rapidly progressive renal failure.
Plasma exchanges, either alone or in combination with steroids and / or immunosuppressants, have been proposed in the most severe forms, as well as intravenous Ig.
Recurrence on the renal transplant:
Nephropathy of rheumatoid purpura may recur on the renal transplant, especially as the transplant is intrafamily.Generally, recurrence is restricted to the kidney and patients do not show any extrarenal signs. The actuarial risk of recidivism is estimated at 35% and graft loss in relation to recurrence at 11% at 5 years. A short delay between renal transplantation and the first clinical signs of the disease is a risk factor to consider and it is usually recommended to respect 1 year of delay before transplanting the patient. The use of ciclosporin does not appear to reduce the risk of recurrence.