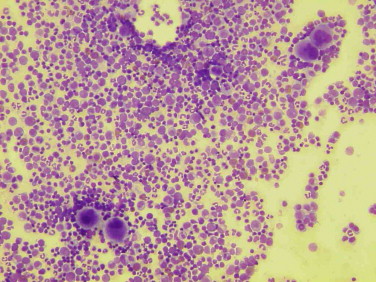
Thrombocytosis is defined as a platelet count greater than 600,000 / mm3. We distinguish secondary thrombocytosis (iron deficiency, inflammation or hyposplenism) of “primitive” thrombocytosis associated with a myeloproliferative disorder.
DIAGNOSTIC CIRCUMSTANCES:
Clinical manifestations of thrombocytosis are almost exclusively in the forms linked to a myeloproliferative disorder.They are dominated by a predisposition to thrombotic events, more typically arterial and venous, or hemorrhagic.This may be an infringement of the great arteries (stroke, myocardial infarction, but also of deep venous thrombosis, including peripheral or portal system), or microcirculatory impairment (digital ischemia, erythromelalgia, defined by feelings of palmar or plantar burns with increased local heat and purplish toes, enhanced by aspirin, livedo).Headaches are common. Two main thrombosis risk factors have been identified in essential thrombocythemia: a history of thrombosis, and age greater than 60 years (15% per year after 60 years as against 1.7% a year before age 40). Hemorrhagic manifestations, mainly mucosal (epistaxis, gastrointestinal bleeding) are related to an impairment of primary hemostasis, and are again the fact of thrombocytosis “primitive”. This thrombopathy (and therefore the risk of bleeding) is directly proportional to the importance of thrombocytosis. Secondary thrombocytosis is exceptionally difficult to thrombotic and / or hemorrhagic.

Thrombocytosis
BALANCE ETIOLOGICAL:
The initial assessment (clinical examination and NFS) can provide the outset of the arguments in favor of a myeloproliferative disorder: splenomegaly, thrombocytosis than 1 million / mm3, érythromyélémie (myelofibrosis), leukocytosis with myelemia (chronic myeloid leukemia – CML), polycythemia (polycythemia vera).
Initially, the etiologic will help eliminate secondary thrombocytosis (Box 1):
– Body-Howell Jolly research on blood smear looking for a hyposplenism, it is functional (celiac disease, amyloidosis, sickle cell disease, sarcoidosis, irradiation) or post-splenectomy;
– Decreased ferritin (iron deficiency);
– Inflammatory balance: ESR, CRP, fibrinogen.
Box 1. secondary thrombocytosis
Splenectomy, functional hyposplenism
Iron deficiency
Inflammatory syndrome:
– Chronic inflammatory diseases (vasculitis, inflammatory bowel disease, rheumatoid arthritis)
– Infections
– Cancers, blood disorders
Transient thrombocytosis:
– Vinca Alkaloids vincristine, vinblastine
– Stress, severe trauma, surgery, bleeding
– Medullary Hyperactivity: hemolytic anemia, exit post-chemotherapy aplasia …
If negativity of etiological, we must move towards a primitive thrombocytosis: essential thrombocythemia, polycythemia vera (or polycythemia vera), chronic myeloid leukemia or myelofibrosis (or idiopathic myelofibrosis) beginner.
It should be noted the existence of thrombocytosis associated with myelodysplastic syndromes with specific karyotypic abnormalities (including a deletion 5q in) important to recognize because that can benefit from specific treatments (lenalidomide). The evidence for thrombocytosis “myeloproliferative” are:
– Symptomatic thrombocytosis (bleeding and / or thrombosis);
– Chips> 1 million / mm3;
– Splenomegaly; leukocytosis or polycythemia associated
– Myelemia or, conversely, cytopenias (myelofibrosis);
– Abnormalities in primary hemostasis (bleeding time, platelet function) associated with a thrombopathy (the platelet aggregability function is often disturbed in myeloproliferative thrombocytosis).
The diagnosis of essential thrombocythemia may ultimately be retained after eliminating other myeloproliferative syndromes, mainly with the help of the following tests:
– Absence of bcr-abl rearrangement (Polymerase Chain Reaction study on peripheral neutrophils) to rule out chronic myelogenous leukemia;
– Normal erythrocyte total, against polycythemia vera the diagnosis;
– No-éythro myelemia or marrow fibrosis on bone marrow biopsy as part of myelofibrosis;
– Giant platelets and / or megakaryocytes large on the myelogram;
– Spontaneous growth of megakaryocytes in the absence of thrombopoietin (the bone marrow aspirate).
Recently, somatic mutation (V617F) affecting the protein tyrosine kinase JAK2 was identifi ed in myeloproliferative disorders not related to BCR-ABL (CML): this activating mutation is present in almost 100% of cases the disease polycythemia vera and in about 50% of cases of myelofibrosis and essential thrombocythemia.
His research must be systematic in all BCR-ABL negative myeloproliferative syndrome. It currently has no treatment may inhibit this protein (imatinib, or Gleevec, a potent inhibitor of tyrosine kinase BCR-ABL in CML treatment validated, are not effective on JAK2).
TREATMENT:
Thrombotic risk in secondary thrombocytosis is considered low, requiring no antiplatelet therapy. However, one can observe higher thrombocytosis 1 000 000 / mm3 transients in splenectomy suites that can lead to a temporary prescription of aspirin.
As part of essential thrombocythemia, the reference treatment is hydroxyurea (Hydrea®), with a goal of less platelets 500,000 or 600,000 depending on the context (thrombotic risk) and haematological tolerance of treatment.Therapeutic alternatives are anagrelide and pipobroman (Vercyte®). The cytoreductive therapy is indicated in situations with high risk of thrombotic events (elderly with a history of thrombosis). The aspirin therapy is recommended in all cases after a first thrombotic event, but his prescription is common in primary prevention in deemed risk situations (after verification of platelet function).
Leave a Reply